Epigenetic Regulation of Aging and its Rejuvenation
Yongpan An 1,2, Qian Wang 3, Ke Gao 1, Chi Zhang 1, Yanan Ouyang 1, Ruixiao Li 1, Zhou Ma 1, Tong Wu 1, Lifan Zhou 1, Zhengwei Xie 3,✉, Rui Zhang 2,✉, Guojun Wu 1,✉
MedComm Open Access. 2025 Sep 1;6(9):e70369. doi: 10.1002/mco2.70369PMCID: PMC12402629 PMID: 40904701
Ronald Peters, MD, MPH – Comments
The epigenome is the chemical modifications to DNA and associated proteins that controls which genes are turned on or off without changing the DNA sequence. This mysterious cellular bio-energy network responds to messages sent from from your body and mind in response to the challenges of life in order to activate an appropriate DNA response. The epigenome is dynamic and can be influenced by factors like diet, stress, lifestyle, and toxins. This allows the body to adjust and respond to its environment. Medical research explores the physiological features of epigenetics but is hesitant to dive into the mystifying world of consciousness and its governing influence on the body. Basically, your DNA is the hardware and the epigenome is the software, which responds moment to moment to your thoughts, emotions and how you live your life.
“When you understand that with every feeling and thought, in every instant, you are performing epigenetic engineering in your own cells, you suddenly have a degree of leverage over your health and happiness that can make a critical difference.” (Dawson Church, The Genie in Your Genes)
ABSTRACT
Aging increases the global burden of disease, yet its molecular basis remains incompletely understood. Recent studies indicate that reversible epigenetic drift—spanning DNA methylation clocks, histone codes, three‐dimensional chromatin, and noncoding RNA networks—constitutes a central regulator of organismal decline and age‐related diseases. How these epigenetic layers interact across different tissues—and how best to translate them into therapeutic strategies—are still open questions. This review outlines the specific mechanisms by which epigenetic changes influence aging, highlighting their impact on genomic instability, stem‐cell exhaustion, and mitochondrial dysfunction. We critically evaluate emerging rejuvenation strategies—partial OSKM reprogramming, CRISPR–dCas9 epigenome editing, NAD⁺/sirtuin boosters, HDAC inhibitors, microbiota transfer, and precision lifestyle interventions—detailing their efficacy in resetting epigenetic age and restoring tissue homeostasis. Integrating single‐cell multiomics and second‐generation epigenetic clocks, we propose a roadmap for translating these insights into safe, personalized antiaging medicine.
Keywords: aging, epigenetic rejuvenation, epigenetic clocks, epigenetic mechanisms, emerging rejuvenation strategies
This article provides an overview of the epigenetic mechanisms and key targets involved in aging, including DNA methylation, histone modifications, chromatin remodeling, and changes in noncoding RNAs. It also introduces various intervention strategies aimed at reversing epigenetic changes to delay aging and treat diseases.
- Introduction
Aging refers to the process by which the functions of various levels of the organism (cells, tissues, organs, etc.) gradually decline and structural changes occur over time. This is typically accompanied by reduced adaptability, weakened immune function, metabolic disorders, and an increased risk of age‐related diseases (such as cardiovascular diseases, cancer, neurodegenerative diseases, etc.), ultimately leading to death [1]. As medical technology has advanced, the global average lifespan has risen markedly. With the drop in the birth rate, populations are aging, and the 65‐plus‐age group is growing faster than others. The United Nations projects that by 2050, one‐sixth of the global population will be over 65 years of age, and those over 80 years will triple. Since aging is a major factor in chronic diseases such as cancer, it has replaced infectious diseases as the main cause of death and disability, imposing a heavy social burden. Thus, research on aging is highly valuable as it can achieve healthy longevity. Studies show that antiaging research may yield more economic benefits than tackling individual diseases [2].
At present, the widely accepted mechanisms of aging mainly include 14 types, such as genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, autophagy dysfunction, deregulated nutrient‐sensing, mitochondrial dysfunction, cellular senescence, and so on [3]. Meanwhile, research shows that compared with genetic information, the loss of epigenetic information plays a core role in the regulation of aging. This conclusion has been confirmed in various model organisms, including yeast, nematodes, fruit flies, and mice [4, 5]. Epigenetics refers to the discipline of heritable changes in gene function without changes in the DNA sequence, which eventually leads to phenotypic changes. As we age, epigenetic changes accumulate, leading to gradual alterations in gene expression and accelerating the aging process. The main mechanisms of these epigenetic changes in regulating longevity pathways include alterations in DNA methylation patterns, abnormal posttranslational modifications of histones, abnormal chromatin remodeling, and dysfunction of noncoding RNAs (ncRNAs). These regulatory and often reversible changes affect gene expression and other cellular processes, leading to the occurrence and progression of various age‐related human pathologies, such as cancer, neurodegenerative diseases, metabolic syndrome, and bone diseases [6].
An increasing number of studies suggest that lifestyle interventions, chemical drug treatments, gene therapy, stem cell transplantation, gut microbiota transfer, immunotherapy, heterochronic parabiosis, and reprogramming are epigenetic therapeutic strategies that can effectively improve or even reverse the aging state of the organism [7, 8]. Therefore, epigenetic regulation can be seen as a crucial entry point for further understanding the mechanisms underlying aging, exploring new aging biomarkers, and developing antiaging drugs and clinical treatment strategies in the future.
The objective of this review is to explore the role of epigenetic mechanisms in aging and their contribution to age‐related diseases. We will examine the key epigenetic processes—DNA methylation, histone modifications, and ncRNAs—and discuss how these mechanisms accumulate over time and influence aging. This review will also highlight recent advances in epigenetic rejuvenation therapies, such as CRISPR/Cas9‐based epigenetic editing, small molecule modulators, and the use of ncRNAs to restore youthful gene expression. These therapies offer promising strategies for extending healthspan and improving the quality of life for aging populations. Additionally, the review will address challenges and risks associated with epigenetic therapies, including off‐target effects and tumorigenesis, and propose future research directions to overcome these issues. By synthesizing recent research, this review aims to provide a comprehensive understanding of the role of epigenetics in aging and disease, and the potential of epigenetic interventions for rejuvenation.
- Epigenetic Mechanisms of Aging
The epigenetic modification mechanisms associated with aging mainly include changes in DNA methylation patterns, abnormal posttranslational modifications of histones, abnormal chromatin remodeling, and dysfunction of ncRNA (Figure 1). This section will systematically elucidate the interplay between epigenetic dysregulation and aging, focusing on the latest discoveries and their implications for aging and disease, while also discussing potential targets and mechanisms that may delay or even reverse the aging process (Figure 2).
FIGURE 1.
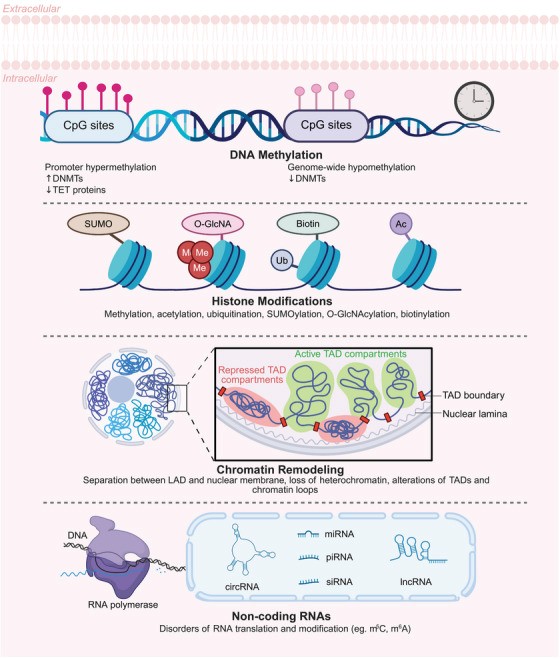
Epigenetic mechanisms underlying the aging process. This schematic diagram summarizes the major epigenetic mechanisms contributing to the aging process, including DNA methylation alterations, histone modifications, chromatin structural changes, and noncoding RNA dysregulation. The illustration shows how promoter hypermethylation silences critical genes while global hypomethylation occurs across the genome, mediated by DNMTs and TET proteins acting on CpG sites. Various histone modifications (methylation, acetylation, ubiquitination, SUMOylation, O‐GlcNAcylation, and biotinylation) dynamically regulate chromatin states. Age‐related chromatin remodeling involves disruption of TAD organization, with changes in active/repressed compartments, boundary integrity, and LAD, accompanied by heterochromatin loss. Additionally, noncoding RNAs (lncRNAs, circRNAs, siRNAs) and RNA modifications exhibit age‐associated dysregulation, affecting transcriptional and translational processes. Collectively, these interconnected epigenetic changes drive the progressive functional decline characteristic of cellular aging. This figure was created by BioRender. Abbreviations: DNMT, DNA methyltransferase; TET, ten‐eleven translocation; CpG, cytosine–phosphate–guanine dinucleotide; SUMO, SUMOylation; O‐GlcNA, O‐GlcNAcylation; TAD, topologically associating domain; LAD, lamina‐associated domain; m5C, 5‐methylcytosine; m6A, N⁶‐methyladenosine.
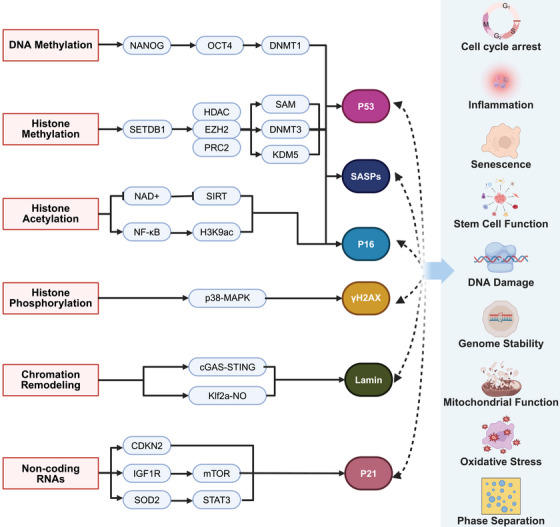
The schematic illustrates key epigenetic mechanisms and their downstream cellular effects. The left section outlines critical epigenetic mechanisms, including DNA methylation (mediated by DNMT1, DNMT3, and associated with pluripotency factors NANOG and OCT4), histone methylation (involving SETDB1, EZH2/PRC2, and KOM5 with H3K9ac and SAM as cofactors), histone acetylation/phosphorylation (featuring H3K9ac, HDAC, and p38–MAPK), and chromatin remodeling (linked to GAS–STING, mTOR, and STAT3 pathways). The right section demonstrates their downstream effects on cellular functions, such as cell cycle regulation and senescence (controlled by p53, p16, CDKN2, and p21), stress responses (including oxidative stress via SOD2, mitochondrial function through NAD+, and DNA damage repair), stem cell dynamics (influenced by NANOG/OCT4 and mTOR signaling), and phase separation in nuclear organization. Together, this integrated network reveals how epigenetic modifications converge with signaling pathways to govern genome stability, inflammation, senescence, and stem cell function. This figure was created by BioRender.
Epigenetic mechanisms underlying the aging process. This schematic diagram summarizes the major epigenetic mechanisms contributing to the aging process, including DNA methylation alterations, histone modifications, chromatin structural changes, and noncoding RNA dysregulation. The illustration shows how promoter hypermethylation silences critical genes while global hypomethylation occurs across the genome, mediated by DNMTs and TET proteins acting on CpG sites. Various histone modifications (methylation, acetylation, ubiquitination, SUMOylation, O‐GlcNAcylation, and biotinylation) dynamically regulate chromatin states. Age‐related chromatin remodeling involves disruption of TAD organization, with changes in active/repressed compartments, boundary integrity, and LAD, accompanied by heterochromatin loss. Additionally, noncoding RNAs (lncRNAs, circRNAs, siRNAs) and RNA modifications exhibit age‐associated dysregulation, affecting transcriptional and translational processes. Collectively, these interconnected epigenetic changes drive the progressive functional decline characteristic of cellular aging. This figure was created by BioRender. Abbreviations: DNMT, DNA methyltransferase; TET, ten‐eleven translocation; CpG, cytosine–phosphate–guanine dinucleotide; SUMO, SUMOylation; O‐GlcNA, O‐GlcNAcylation; TAD, topologically associating domain; LAD, lamina‐associated domain; m5C, 5‐methylcytosine; m6A, N⁶‐methyladenosine.
FIGURE 2.
The schematic illustrates key epigenetic mechanisms and their downstream cellular effects. The left section outlines critical epigenetic mechanisms, including DNA methylation (mediated by DNMT1, DNMT3, and associated with pluripotency factors NANOG and OCT4), histone methylation (involving SETDB1, EZH2/PRC2, and KOM5 with H3K9ac and SAM as cofactors), histone acetylation/phosphorylation (featuring H3K9ac, HDAC, and p38–MAPK), and chromatin remodeling (linked to GAS–STING, mTOR, and STAT3 pathways). The right section demonstrates their downstream effects on cellular functions, such as cell cycle regulation and senescence (controlled by p53, p16, CDKN2, and p21), stress responses (including oxidative stress via SOD2, mitochondrial function through NAD+, and DNA damage repair), stem cell dynamics (influenced by NANOG/OCT4 and mTOR signaling), and phase separation in nuclear organization. Together, this integrated network reveals how epigenetic modifications converge with signaling pathways to govern genome stability, inflammation, senescence, and stem cell function. This figure was created by BioRender.
2.1. DNA Methylation Dynamics
DNA methylation is one of the most well‐known epigenetic modifications. DNA methylation is catalyzed by DNA methyltransferases (DNMTs) using S‐adenosyl‐methionine (SAM) as the methyl donor, which transfers a methyl group (–CH3) to specific bases in defined genomic regions. In mammals, the three principal methyltransferases are DNMT3A, DNMT3B, and DNMT1. According to the methylation pattern they establish, these enzymes are classified as either de novo or maintenance methyltransferases. DNMT3A and DNMT3B are de novo methyltransferases responsible for establishing methylation on previously unmethylated DNA. This de novo methylation—that is, the creation of new methylation marks on unmethylated cytosines—occurs predominantly during embryonic stem‐cell development and sets up novel methylation patterns. DNMT1 is a maintenance methyltransferase that preserves existing methylation patterns. After DNA replication, DNMT1, as an integral component of the replication complex, recognizes hemimethylated CpG sites on the nascent strand. Through a nucleophilic attack, it catalyzes methylation of these hemimethylated positions, thereby copying the methylation pattern from the parental strand to the daughter strand and ensuring faithful propagation of the methylation landscape originally established by the de novo methyltransferases [9, 10]. This modification is crucial for regulating gene expression, maintaining genomic stability, and facilitating cellular differentiation.
2.1.1. Age‐Related Methylation Patterns
Age‐related changes in DNA methylation patterns are not only widespread but also have a significant impact on gene expression, influencing various life processes such as metabolism, inflammation, cancer, cardiovascular diseases, neurological disorders, and aging [11, 12, 13, 14, 15, 16]. Early studies found that in certain aging human and mouse cells, tissues, and organs (e.g., T cells, small intestine mucosa, liver, and brain), there is often a decrease in overall DNA methylation levels [9], while focal hypermethylation can emerge [17, 18] and correlate with a higher incidence of cardiovascular disease in older individuals [15]. A meta‐analysis from 128 mammal species revealed that hypermethylation of CpG islands is present in aging blood, brain, cortex, liver, muscle, and skin [19]. These patterns arise primarily from cell‐type‐specific, age‐stage‐specific, and stress‐signal‐specific transcriptional regulation of the three canonical DNMTs (DNMT1, DNMT3A, and DNMT3B). In replicative‐exhaustion cellular models, all three DNMTs are downregulated. By contrast, during in vivo aging driven by oxidative stress or chronic inflammation, DNMT1 is suppressed via the telomere–p53 axis, whereas DNMT3A/3B are upregulated through NF‐κB/STAT3 activation. The combined effect is the emergence of an “aging epigenetic signature” characterized by global hypomethylation superimposed on region‐specific hypermethylation [20, 21, 22]. Current research is increasingly focused on age‐related, region‐or site‐specific methylation changes—such as differentially methylated regions and differentially methylated positions—to dissect the molecular underpinnings of this epigenetic drift [23].
DNA methylation regulates aging by silencing or inducing aging‐related genes. During aging, the hypermethylation of specific gene promoters can silence crucial genes, particularly those involved in tumor suppression and immune response. For example, studies have shown that hypermethylation of the TP53 gene is associated with reduced expression in older populations [24, 25], which may contribute to the increased incidence of malignancies in the aging population. Furthermore, hypermethylation of inflammatory genes such as IL1β, IL6, and TNFα is linked to atherosclerosis [26], which poses significant health risks to older adults. Reduced DNA 5mC levels can upregulate PSG, endogenous retroviruses (ERVs), p15, p16, p21, and LINE‐1, while downregulating ELOVL2. Increased DNA 6mA can upregulate heat stress response genes. These changes can induce senescence‐associated secretory phenotype (SASP), inhibit the cell cycle, and accelerate cellular senescence [27, 28, 29, 30, 31, 32].
Another mechanism of DNA methylation in aging suggests that the widespread decay of the methylome reflects an “epigenetic maintenance system” that supports development, cell differentiation, and cell identity maintenance. Changes in methylation at specific genomic sites are crucial for preserving stem cell identity and function [9]. DNA methylation changes often reduce stem cell numbers and functions, such as impairing self‐renewal and causing differentiation bias, mirroring what is observed in aging [33]. Supporting this, single‐cell analysis in mice shows that liver cells’ epigenetic aging is precisely tracked, while muscle stem cells show minimal epigenetic age changes [34]. This indicates that the epigenetic clock responds when stem cells are stimulated to divide [35]. In summary, the mechanisms of how methylation changes accelerate aging are currently thought to be primarily through the silencing or induction of aging‐related genes and the impact on stem cell numbers and functions.
2.1.2. Epigenetic Clocks
Epigenetic clocks are DNA methylation‐based biomarkers used to estimate the difference between an individual’s biological age and chronological age. The core concept of epigenetic clocks is that over time, the DNA methylation patterns at certain specific locations in the genome undergo predictable changes, which are closely related to aging, health status, and lifespan prediction. Different epigenetic clock models rely on different algorithms and DNA methylation sites, and various types of age clocks have been developed with different machine algorithms, research species, and tissue sources [12, 36, 37, 38, 39].
The first‐generation DNA methylation clocks (DNAm) estimate biological age using specific CpG sites, with the most famous being the Horvath clock and the Hannum clock [40, 41]. Second‐generation clocks, such as PhenoAge, GrimAge, and DunedinPACE [42, 43, 44], incorporate clinical biomarkers alongside DNA methylation data, improving accuracy and applicability. As a result, these clocks are not only associated with physiological age but also predict the onset of aging‐related diseases, facilitate disease diagnosis, and contribute to health assessments [12, 36, 37, 39, 45].
With the emergence of various machine learning and deep learning models, the algorithms for DNAm are continuously being updated. For example, the DNAge model, based on Horvath’s pan‐tissue clock, assesses the actual age of skeletal muscle in aging mice through exercise [46]. Recent research has focused more on lifestyle and environmental factors, such as alcohol consumption and sleep quality [47], and their impact on epigenetic age acceleration (EAA) assessments. For example, the newly developed GrimAge V2, using deep learning methods, shows better predictive performance than its V1 version [48].
The clinical application value of epigenetic clocks is becoming increasingly prominent. Studies have shown that EAA assessed by epigenetic clocks is associated with an increased risk of aging‐related diseases, such as Alzheimer’s disease (AD) and cardiovascular diseases [45, 49, 50]. One study utilized DNA methylation data from 378 women to develop the first second‐generation epigenetic age clock for skin, which can accurately predict skin aging phenotypes represented by wrinkle grade, visual facial age, and visual age progression [51].
Furthermore, these clocks have primarily been developed for European or Hispanic populations. A study using DNA methylation data from blood samples of Koreans demonstrated the applicability of epigenetic clocks in East Asian populations. This study not only included chronic disease factors, blood biomarker levels, and lung function but also considered health behavior factors, socioeconomic status, and psychological stress levels [52], confirming the association between EAA and environmental factors in Asian populations.
2.2. Histone Modification Alterations
Histones are small, basic proteins rich in amino acids such as lysine and arginine, and they are the major structural proteins of eukaryotic chromatin [53]. They bind to DNA to form nucleosomes, which are the fundamental repeating units of chromatin. There are four core histones: H2A, H2B, H3, and H4, each composed of two molecules to form an octamer, around which DNA is wound to form the nucleosome [54]. H1 is the linker histone [55], located at the linker DNA regions between nucleosomes, helping to compact higher‐order chromatin structures. The amino acid sequences of histones are highly conserved through evolution, especially H3 and H4, indicating the importance of their functions [56].
Histone modification refers to the process by which specific amino acid residues (especially at the N‐terminal tails) on histones undergo covalent chemical modifications. By adding or removing small chemical groups (such as methyl, acetyl, or phosphate groups), histone modifications dynamically regulate chromatin structure and gene expression [22, 57]. These modifications do not alter the DNA sequence but are heritable and influence cellular functions, making them one of the core mechanisms of epigenetic regulation. This process is dynamic and reversible, with “writers” (such as histone acetyltransferases [HATs] and HMTs) adding modifications and “erasers” (such as histone deacetylases [HDACs] and KDMs) removing them [58, 59]. Histone modifications include various types, with common ones being methylation, acetylation, phosphorylation, and ubiquitination. On one hand, these posttranslational modifications can activate or repress gene expression by regulating chromatin structure. On the other hand, different combinations of modifications can form a “histone code” [60, 61], which is recognized by effector proteins (such as proteins containing Bromo/Chromo domains) and recruits the transcriptional machinery to initiate distinct gene expression programs. Histone modifications play multiple biological roles in eukaryotic cells, participating in the regulation of chromatin structure, gene expression, DNA damage and repair, and the cell cycle [60, 61]. These functions are realized through complex interaction networks, and different modifications may have synergistic or antagonistic effects [62, 63]. Abnormal histone modifications are closely associated with various diseases, such as tumors [64], neurodegenerative diseases [65], and aging [66, 67, 68], and are potential therapeutic targets.
2.2.1. Histone Methylation
Recent studies have shown that histone methylation marks such as H3K4me3, H3K27me3, and H3K36me3 undergo significant changes during aging, and these changes directly affect the aging and repair capacity of cells [69]. H3K4me3 is closely related to the expression of aging‐associated genes. Research has found that in the hematopoietic stem cells (HSCs) of aging mice, the level of H3K4me3 is increased [70], while in physiologically aged human HSCs, H3K4me1, H3K4me3, and H3K27ac levels are reduced [71]. Histone methylation exhibits significant spatiotemporal‐specific changes during aging, and these modifications directly impact chromatin structure and gene expression patterns through epigenetic regulatory networks, becoming a key molecular basis for the development of aging and related diseases [72, 73, 74, 75]. Recent studies have shown that aging cells display typical characteristics of methylation reprogramming: (1) Repressive marks, such as H3K27me3 (trimethylation of lysine 27 on histone H3), undergo specific loss at gene promoter regions, especially at cell cycle inhibitor genes (such as p16INK4a/CDKN2A) [76, 77, 78, 79]. Similar to the mechanism of DNA hypomethylation in promoting aging, histone hypo‐methylation, such as decreased H3K9me3, H4K20me3, H3K9, and H3K36 methylation, can upregulate PSG, p15, p16, p21, and LINE‐1, while downregulating ELOVL2 [27, 28, 29, 30, 31, 32]. These changes induce cell cycle arrest and activate SASP. (2) The reduction of H3K9me3 in heterochromatic regions leads to decreased genomic stability, abnormal activation of transposons, and nuclear structure disruption [31, 80, 81]. (3) The abnormal accumulation of the active mark H3K4me3 at the promoter regions of metabolism‐related genes [28, 57, 82], which reprograms cellular. These changes are closely associated with the dysregulation of methylation‐modifying enzymes, including the reduced activity of the Polycomb repressive complex 2 (PRC2) core enzyme EZH2 [83, 84], the upregulation of KDM demethylase enzymes (such as KDM5A/B) [63, 69, 85], and the age‐dependent reduction of the methyl donor SAM [86, 87, 88, 89, 90, 91]. These changes induce DNA damage and oxidative stress, activate mTOR to regulate metabolism, and promote aging.
In aging‐related diseases, these methylation abnormalities exhibit tissue‐specific patterns. In neurodegenerative diseases (such as AD), neurons in the prefrontal cortex show a significant increase in H3K9me2 [92] and a decrease in H3K27me3 [93], leading to the silencing of synaptic plasticity genes [94]; in muscle atrophy, aging mice muscles show an elevated level of H3K27me3 [95, 96], which inhibits the differentiation of muscle stem cells and muscle regeneration. The expression of the antiaging protein Klotho can suppress the activity of the H3K27 demethylase KDM6B/JMJD3, reduce H3K27me3 levels, and promote muscle stem cell differentiation [97]. Additionally, H3K36me3 levels significantly decrease, which may be related to a decline in DNA repair capacity [98] and impaired muscle stem cell function [99]; in cardiovascular aging, the loss of H3K27me3 in endothelial cells promotes the expression of inflammatory factors, accelerating the progression of atherosclerosis [100, 101, 102, 103]; in aging‐related cancers, the abnormal distribution of H3K36me3 leads to DNA damage repair defects and increased genomic instability [104, 105, 106]. Notably, these epigenetic changes form a positive feedback loop with classic aging markers (such as mitochondrial dysfunction and stem cell depletion): increased mitochondrial reactive oxygen species (ROS) production can suppress the activity of HMTs [107, 108], while abnormal methylation patterns further affect nuclear–mitochondrial communication [109].
Currently, therapeutic strategies targeting these methylation regulatory mechanisms (such as EZH2 inhibitors, KDM inhibitors, and methyl donor supplementation) have shown potential for improvement in various aging‐related disease models [109, 110], providing important directions for the development of new antiaging interventions.
2.2.2. Histone Acetylation
Recent studies have revealed the bidirectional regulatory characteristics of histone acetylation in the aging process: on one hand, there is a progressive decline in global acetylation levels, while on the other hand, specific functional genomic regions (such as inflammation‐related gene loci) show abnormal acetylation accumulation. This “global loss‐local gain” pattern constitutes an epigenetic hallmark of aging [3, 97, 111]. At the molecular level, aging‐related histone acetylation disorders mainly involve three core aspects: imbalance in the acetylation “writing–erasing” system, disruption of the metabolic–epigenetic network, and changes in chromatin spatial organization [112, 113, 114].
The dynamic equilibrium system composed of HATs and HDACs undergoes significant changes during aging [115]. Studies have shown that the expression and catalytic activity of major HATs such as p300/CBP decrease with age, and their nuclear localization also becomes abnormal [116, 117]. This functional decline of HATs leads to a reduction in acetylation levels at several key sites, including H3K14 and H4K16 [118, 119]. Meanwhile, the expression and activity of NAD+ dependent class III HDACs (sirtuin family) show tissue‐specific variations, SIRTs expression is downregulated in most tissues [120, 121, 122, 123]. Notably, the functional decline of SIRT6 in the aging process is the most significant, leading to the abnormal accumulation of its target sites H3K9ac and H3K56ac [24, 124, 125], which in turn affects DNA damage repair and genomic stability.
Histone acetylation is closely linked to cellular metabolic states, and this feature is particularly prominent in the aging process. The decline in mitochondrial function results in reduced acetyl‐CoA production, which is the primary cause of the global decline in acetylation [126, 127]. On the other hand, age‐dependent reductions in NAD+ levels weaken the activity of SIRT1/6, forming a “metabolic–epigenetic vicious cycle”: decreased NAD+ → reduced SIRT activity → high histone acetylation → proinflammatory gene expression → mitochondrial damage → further depletion of NAD+ [120, 128, 129]. Additionally, aging‐related changes in α‐ketoglutarate levels indirectly regulate acetylation patterns by affecting TET enzyme activity [130].
The changes in histone acetylation during aging significantly impact the higher‐order structure of chromatin. Topology‐associated domains (TADs) boundary integrity of euchromatic regions (marked by H3K27ac) is disrupted in aged cells, leading to abnormal interactions of previously isolated functional genomic regions [131]. This structural change is closely related to the rearrangement of lamina‐associated domains (LADs), manifesting as the abnormal accumulation of H4K16ac in perinuclear heterochromatin regions [132, 133]. Notably, an “acetylation phase separation abnormality” phenomenon is observed in aging cells: certain transcription factors (such as NF‐κB) form biomolecular condensates by recognizing specific acetylation marks (such as H3K9ac), which exacerbates chronic inflammation [134].
Abnormal histone acetylation is closely associated with the development of various aging‐related diseases. In neurodegenerative diseases, hippocampal neurons show specific loss of H4K12ac, leading to the silencing of synaptic plasticity‐related genes (such as BDNF, Arc) [135]. Single‐cell epigenomic analysis reveals that the abnormal distribution of H3K27ac in neurons of AD patients precedes amyloid plaque formation [136, 137], suggesting that it may serve as an early diagnostic marker. In the cardiovascular system, the abnormal increase in H3K9ac in endothelial cells promotes atherosclerotic plaque formation by activating the NF‐κB signaling pathway [138, 139]. In metabolic diseases, the reprogramming of H3K27ac in liver tissue directly leads to the abnormal expression of key gluconeogenesis enzymes (PEPCK, G6Pase), exacerbating age‐related glucose metabolism disorders [140].
Intervention strategies targeting histone acetylation regulation in aging primarily focus on three directions: metabolic reprogramming (such as NAD+ precursor supplementation) [141, 142, 143], epigenetic editing (such as HATs/HDACs targeting regulation) [144], and anti‐inflammatory treatment (such as SASP inhibitors) [145]. Preclinical studies have shown that nicotinamide mononucleotide (NMN) can restore SIRT1 activity by elevating NAD+ levels [146]. Selective HDAC inhibitors (HDACis) have shown protective effects in neurodegenerative disease models [147]. Future research needs to address challenges such as tissue‐specific delivery and long‐term safety, and explore precision intervention strategies based on single‐cell epigenomics.
2.2.3. Histone Phosphorylation
Recent studies have found that histone phosphorylation in aging cells exhibits significant reprogramming characteristics, manifested as abnormal accumulation of DNA damage‐related phosphorylation marks and reduced efficiency of signal‐responsive phosphorylation events. At the molecular level, aging‐associated histone phosphorylation disorders mainly involve three core aspects: (1) the functional decline of the DNA damage response (DDR) system leading to the persistent deposition of γ‐H2AX (H2AXS139ph); (2) changes in the activity of signaling pathways such as mitogen‐activated protein kinases (MAPK) and Aurora kinases, causing a remodeling of phosphorylation patterns; (3) disruption of the phosphatase network, which impedes dephosphorylation processes [65, 148, 149, 150, 151].
In young cells, DNA double‐strand breaks (DSBs) rapidly induce phosphorylation at the C‐terminal S139 site of H2AX (forming γ‐H2AX), which serves as a damage marker to recruit repair factors [148, 152]. However, aging cells exhibit two characteristic changes: one is a significant increase in baseline γ‐H2AX levels, indicating increased genomic instability [153]; the other is a decreased ability to form new γ‐H2AX foci upon damage, reflecting reduced repair efficiency [149]. This “high background‐low response” pattern is closely related to decreased ataxia‐telangiectasia mutated (ATM)/ataxia telangiectasia and Rad3‐related protein (ATR) kinase activity and overexpression of PP2A phosphatase [154]. Notably, the abnormal accumulation of γ‐H2AX in aging cells not only marks DNA damage but may also inhibit transcription factor access through spatial hindrance mechanisms, leading to the silencing of important metabolic genes (such as SIRT6) [155].The MAPK and Aurora kinase pathways are key hubs regulating aging‐related phosphorylation. p38–MAPK remains persistently activated in aging cells, which drives the production of the SASP by promoting NF‐κB recruitment [106]. In contrast, the activity of Aurora B kinase declines with age, causing chromosome segregation errors and karyotype instability [156].
2.3. Chromatin Remodeling and Architecture Shifts
Recent studies have shown that aging is accompanied by significant chromatin structural remodeling, including the loss of heterochromatin, disintegration of the 3D genome, and activation of transposons, which are epigenetic changes [157, 158, 159, 160]. The downregulation of nuclear lamina protein Lamin B1 in aging cells leads to nuclear membrane wrinkling and the redistribution of LADs, with LADs dissociating in aged tissues, resulting in the abnormal expression of previously silenced genes [161]. This change is closely associated with the SASP, LAD dissociation releases ERVs, driving chronic inflammation by activating the cGAS–STING pathway [162].
At the level of heterochromatin, several studies have reported aging‐related histone modification abnormalities. SUV39H1 catalyzes the trimethylation of histone H3 at lysine 9 (H3K9me3), which is specifically recognized and bound by the chromodomain of HP1 proteins. HP1 then homodimerizes and recruits additional SUV39H1, driving the formation of liquid‐like heterochromatin droplets and leading to heterochromatin relaxation [163, 164, 165, 166]. During stem cell aging, SUV39H1‐mediated H3K9me3 significantly decreased, leading to the derepression of satellite DNA repeats [167, 168, 169]. In 2023, research indicated that loss of H3K27me3 reactivates LINE‐1 transposons, promoting genomic instability [170]. 3D genome analysis revealed that in aging cells, the transition between A/B compartments increases, TAD boundary integrity weakens (especially at tumor suppressor gene loci such as p53), and enhancer–promoter interactions decrease, all of which collectively lead to disrupted gene expression networks [171, 172].
At the molecular level, chromatin remodeling involves abnormalities in multiple layers of regulation. Dysregulation of histone‐modifying enzymes is manifested by decreased HDAC activity, causing an increase in H4K16ac [173], decreased DNMT expression leading to global hypomethylation [174, 175], and NSD2 upregulation drives the aberrant accumulation of H3K36me2 [176]. This increase renders chromatin more open, facilitating the binding of transcription factors and other proteins to DNA and thereby enhancing gene transcription. Concurrently, the elevated H3K36me2 level mediated by NSD2 antagonizes EZH2‐catalyzed H3K27me3 deposition. Because H3K27me3 is a repressive mark, its reduction weakens chromatin‐mediated repression [177, 178, 179]. The function of chromatin remodeling complexes is also significantly impaired, SWI/SNF engages nucleosomes, transiently disrupts their DNA contacts, and generates short DNA loops that allow the nucleosome to slide to new positions, thereby promoting either transcriptional activation or repression. Concurrently, SWI/SNF forms a dynamic balance with Polycomb complexes (PRC1 and PRC2) during developmental gene regulation. SWI/SNF opens chromatin by dismantling PRC2‐mediated H3K27me3 domains, whereas PRC1 recondenses heterochromatin through deposition of H2AK119ub1. Disruption of this equilibrium is frequently associated with disease [180, 181, 182, 183], These changes drive Brg1 (SMARCA4) to bind the promoter of Klf2a, activating the Klf2a–NO pathway and thereby disrupting stem cell function [184]. Deficiency in the NuRD complex reduces DNA repair efficiency [185, 186, 187]. The regulatory role of ncRNAs is increasingly recognized, with the accumulation of aging‐related long ncRNAs (lncRNAs) like TERRA causing telomere heterochromatin disintegration [188, 189], and the circRNA affecting chromatin accessibility by influencing HMGB1 [190].
These chromatin changes are closely linked to various aging‐related diseases. In neurodegenerative diseases, AD patients’ neurons show an abnormal increase in H3K27ac, promoting tau overexpression [191, 192], while Parkinson’s disease (PD) is associated with increased chromatin accessibility at the SNCA gene locus [193, 194]. Research in cancer shows that aging‐related disruption of TAD boundaries in leukemia activates proto‐oncogenes [195]. Metabolic diseases like diabetes are related to a decrease in enhancer–promoter interactions at the PDX1 locus in pancreatic β‐cells [196], while atherosclerosis involves inflammation triggered by the loss of H3K9me3 in endothelial cells [197].
In response to these findings, researchers have developed various intervention strategies. In epigenetic reprogramming, OSKM induction can partially restore chromatin structure in aging cells [198, 199], and HDACis can improve cognitive function in aging mice [200]. Attempts to target chromatin remodeling include inhibiting NSD2 to delay stem cell aging [201] and activating SIRT6 to enhance heterochromatin stability [202, 203]. In gene therapy, CRISPR–dead Cas9 (dCas9)‐mediated epigenetic editing has achieved precise regulation of disease‐associated loci [204, 205]. Despite the progress, the field still faces challenges such as unclear tissue‐specific regulations and the limitations of 3D genome dynamic monitoring technologies. Future work will need to develop single‐cell multiomics technologies and strengthen clinical translation research.