NAD+ Metabolism in Cardiac Health, Aging, and Disease
Mahmoud Abdellatif, MD, PhD; Simon Sedej, PhD; Guido Kroemer, MD, PhD
Circulation, November 2021
ABSTRACT
Nicotinamide adenine dinucleotide (NAD+) is a central metabolite involved in energy and redox homeostasis as well as in DNA repair and protein deacetylation reactions. Pharmacological or genetic inhibition of NAD+-degrading enzymes, external supplementation of NAD+ precursors, and transgenic overexpression of NAD+-generating enzymes have wide positive effects on metabolic health and age-associated diseases. NAD+ pools tend to decline with normal aging, obesity, and hypertension, which are all major risk factors for cardiovascular disease, and NAD+ replenishment extends healthspan, avoids metabolic syndrome, and reduces blood pressure in preclinical models. In addition, experimental elevation of NAD+ improves atherosclerosis, ischemic, diabetic, arrhythmogenic, hypertrophic, or dilated cardiomyopathies, as well as different modalities of heart failure. Here, we critically discuss cardiomyocyte-specific circuitries of NAD+ metabolism, comparatively evaluate distinct NAD+ precursors for their preclinical efficacy, and raise outstanding questions on the optimal design of clinical trials in which NAD+ replenishment or supraphysiological NAD+ elevations are assessed for the prevention or treatment of major cardiac diseases. We surmise that patients with hitherto intractable cardiac diseases such as heart failure with preserved ejection fraction may profit from the administration of NAD+ precursors. The development of such NAD+-centered treatments will rely on technological and conceptual progress on the fine regulation of NAD+ metabolism.
Nicotinamide adenine dinucleotide (NAD) is essential for the metabolism of eukaryotic cells. The capacity of NAD to shuttle electrons between its oxidized (NAD+) and reduced (NADH) forms is indis-pensable for oxidation-reduction reactions that capture or liberate cellular energy in the form of ATP. Beyond its role in energy metabolism, NAD+ has also been recog-nized as a pivotal signaling molecule and a rate-limiting substrate of multiple enzymes involved in DNA repair, epigenetic regulation, posttranslational modifications, and metabolic adaptation to changing nutritional states.1 Over the period of the last decade, a growing reper-toire of studies transformed our understanding of NAD+ biology and its pathophysiological implications.2,3 In this regard, experimental strategies for NAD+ repletion can delay several hallmarks of aging and simultaneously suppress the manifestation of age-related diseases in rodent models.4–7 On the basis of these observations, NAD+ precursors harbor promise as antiaging drugs igniting renewed interest in the metabolism and pleiotro-pic action of NAD+. However, in spite of the accumulating preclinical evidence in favor of the broad health-improv-ing effects of NAD+ enhancers,8–10 only few clinical trials have been performed in humans.
In the context of cardiovascular morbidity, emerging preclinical evidence indicates that increasing cellular NAD+ content might represent a promising therapeu-tic avenue.11–13 In support of this idea, disrupted NAD+ metabolism is increasingly considered as an amendable cardiovascular risk factor.6,14 In fact, the pathogenesis of various chronic cardiovascular diseases has been consistently shown to coincide with perturbations of NAD+ homeostasis.15–17 The cardiovascular system is particularly vulnerable to such dysregulation in NAD+ metabolism because of the high energy demand of the heart.18 Specifically, depletion of intracellular NAD+ impairs mitochondrial fatty acid β-oxidation and oxida-tive phosphorylation, underscoring that adequate NAD+ availability is critical for the maintenance of myocardial bioenergetic efficiency and, thus, normal pump func-tion. Diminution of the cardiovascular NAD+ pool below a critical threshold also entails major dysfunctions at the cellular level. These include, but are not limited to, deregulated nutrient sensing, epigenetic and gene dysregulation, autophagy impairment, and low-grade inflammation, all of which can independently fuel the development of cardiovascular disease.1
In this in-depth review, we discuss the current under-standing of NAD+ metabolism, and how impaired NAD+ homeostasis aggravates common cardiometabolic risk factors, thus favoring cardiovascular morbidity. We then delve into the therapeutic potential of different NAD+-based therapies against prevalent cardiovas-cular maladies, ranging from ischemic, hypertrophic, arrhythmogenic, diabetic, and dilated cardiomyopathies to heart failure. Last, we present unfolding clinical evi-dence in favor of the utility of NAD+ precursors in car-diovascular medicine.
NAD+ METABOLISM IN THE HEART AND CIRCULATION
Cardiomyocytes accumulate NAD+ mostly within their mitochondria,19 where the bulk of cellular oxidation-re-duction reactions occur. However, NAD+ is also present in the cytosol and nucleus, where NAD+-derived metabo-lites and NAD+-dependent enzymes contribute to various cellular functions.1 In recent years, significant progress has been made in the understanding of tissue-specific NAD+ synthesis, transport, and catabolism.
NAD+ Biosynthesis
Although the liver and, to a lesser extent, the kidney can synthesize NAD+ from the amino acid tryptophan through the kynurenine pathway, the majority of organs, including the heart, lack the enzymes necessary for the de novo biosynthesis of NAD+ (Figure 1A). Instead, cardiac cells generate NAD+ from preformed pyridine moieties such as nicotinamide. Nicotinamide is intracellularly available as an end-product of nonoxidative NAD+ catabolism and, thus, represents a readily available substrate for NAD+ production by NAMPT (nicotinamide phosphoribosyl-transferase), the main rate-limiting enzyme in the NAD+ salvage pathway (Figure 2). However, intracellular recy-cling of NAD+ is not unlimited, because nicotinamide is also regularly metabolized and excreted in urine. There-fore, dietary intake of NAD+ precursors, such as nico-tinamide, nicotinic acid (NA), and nicotinamide riboside (NR)—collectively known as vitamin B3—is required to sustain organismal NAD+ homeostasis. These NAD+ pre-cursors are intracellularly converted to NAD+ through the amidated or deamidated pathways (Figure 2). Explicitly, NAMPT and NMRKs (NR kinases) convert nicotinamide and NR, respectively, into nicotinamide mononucleotide (NMN) via the amidated pathway, whereas NA enters the deamidated pathway to form NA mononucleotide. Both NMN and NA mononucleotide are subsequently used as substrates for NAD+ production in the reaction catalyzed by NMN adenylytransferases.
Different NAD+ precursors might vary in their efficacy to replenish NAD+, both in a tissue- and context-dependent manner. With respect to the human heart, gene expression data (Figure 1A) indicate that NAD+ biosynthetic enzymes of the amidated pathway are much more abundant than those of the deamidated pathway. Indeed, the amidated pathway accounts for 99.3% of cardiac NAD+ stores.20 The protein expression of NMRKs and NAMPT appears to be highly context-dependent. Under physiological conditions, NMRKs are not detectable by protein immunoblotting,21 suggesting that NAMPT might be the sole rate-limiting enzyme for NAD+ biosynthesis expressed in the healthy heart. Hence, it is conceivable that nicotinamide is the primary source of cardiac NAD+ under physiological con-ditions, especially because nicotinamide is the most abun-dant NAD+ precursor in the circulation (2000 nmol/L of nicotinamide versus 7 nmol/L of NR).21,22 In support of this notion, acute NR administration fails to increase cardiac NAD+ content in healthy mice, contrasting with its ability to enhance hepatic NAD+ levels.23 By contrast, dilated car-diomyopathy (DCM) is associated with reduced NAMPT expression and NMRK upregulation, and thus, NR effec-tively replenishes cardiac NAD+.16,24,25
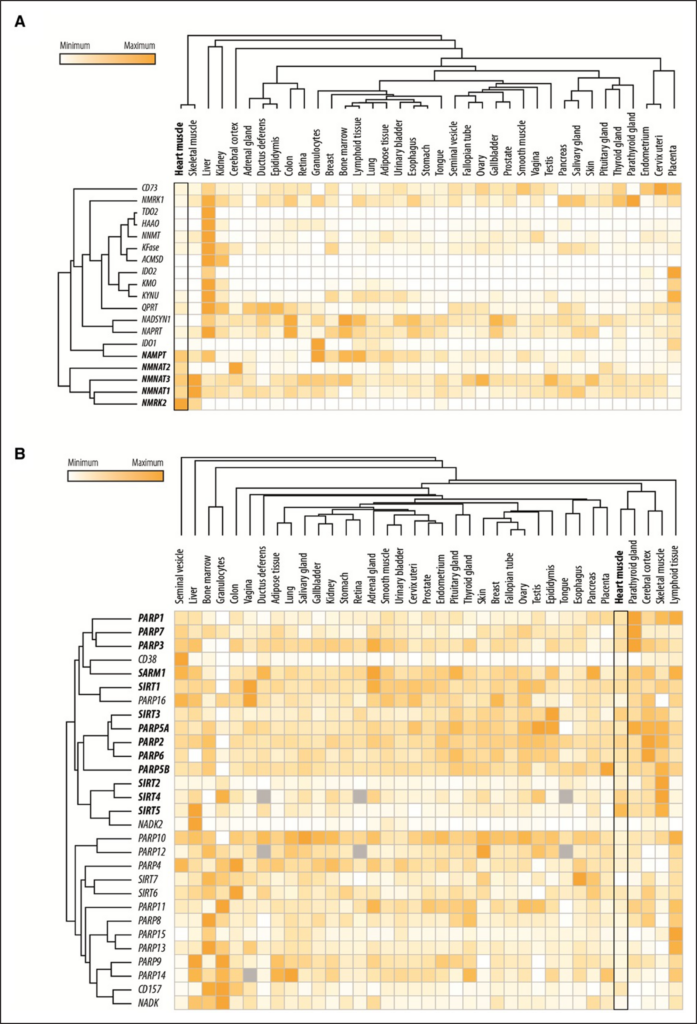
Figure 1. Gene expression levels of the enzymes involved in NAD+ metabolism. Shown are the relative gene expression levels of the enzymes involved in (A) the deamidated and amidated pathways of NAD+ biosynthesis as well as (B) those involved in NAD+ catabolism in the heart and other organs/tissues. The data were retrieved from The Human Protein Atlas (http://www.proteinatlas.org/) and subsequently subjected to hierarchical clustering in Morpheus (https://software.broadinstitute.org/morpheus). Data not available in the atlas are represented by gray boxes. NAD+ indicates nicotinamide adenine dinucleotide, oxidized form.
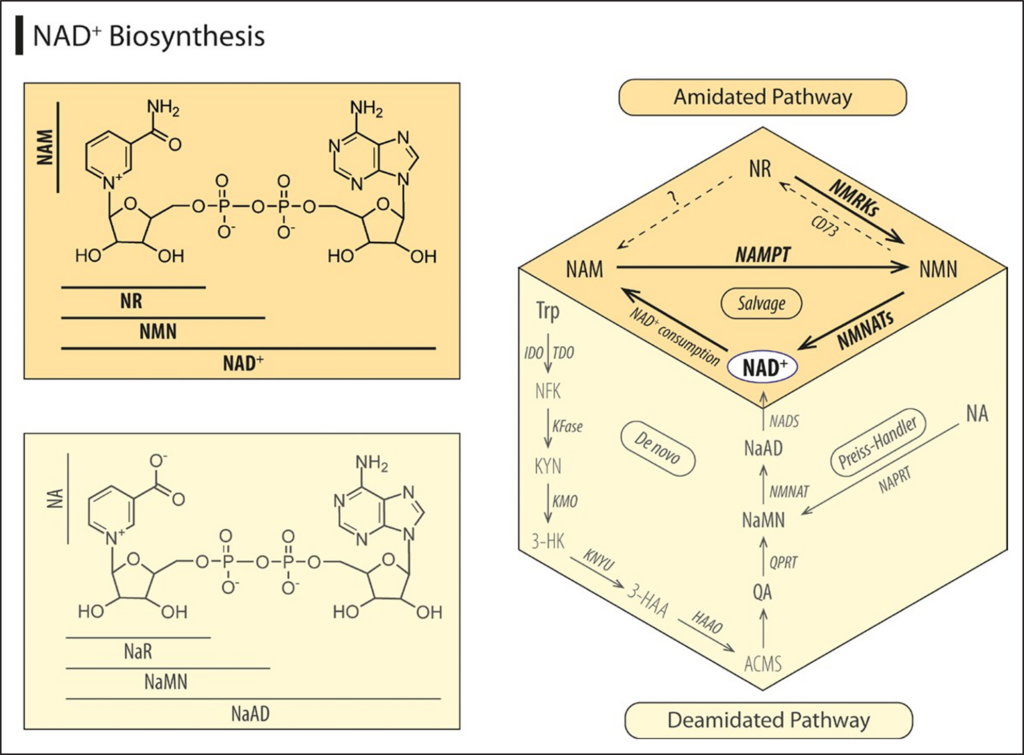
Figure 2. Biosynthetic pathways of NAD+. Because the heart lacks the enzymes necessary for the de novo biosynthesis of NAD+ from the amino acid tryptophan (Trp), cardiac cells instead salvage NAD+ from preformed pyridine moieties, such as nicotinamide (NAM), nicotinic acid (NA), or nicotinamide riboside (NR)—collectively known as vitamin B3. These NAD+ precursors are metabolized to NAD+ through the amidated pathway, where NAM phosphoribosyltransferase (NAMPT) and NR kinases (NMRKs) convert NAM and NR, respectively, into nicotinamide mononucleotide (NMN), whereas NA is introduced to the Preiss-Handler pathway to form NA mononucleotide (NaMN). Both NMN and NaMN are subsequently used as substrates for NAD+ production in the reaction catalyzed by NMN adenylytransferase (NMNAT). Highlighted in dark color are the enzymes and precursors that are sufficiently expressed in the heart (Figure 1A). 3-HAA indicates 3-hydroxyanthranilic acid; 3-HK, 3-hydroxykynurenine; ACMS, 2-amino-3-carboxymuconate-6-semialdehyde; CD73, ecto-5′-nucleotidase; HAAO, 3-hydroxyanthranilic acid dioxygenase; IDO, indoleamine-2,3- dioxygenase; KFase, kynurenine formamidase; KMO, kynurenine 3-monooxygenase; KYN, kynurenine; KYNU, kynureninase; NAAD, NA adenine dinucleotide; NAD+, nicotinamide adenine dinucleotide, oxidized form; NADS, NAD synthetase; NAPRT, NA phosphoribosyltransferase; NFK, N-formylkynurenine; QA, quinolinic acid; QPRT, quinolinate phosphoribosyltransferase; and TDO, tryptophan-2,3-dioxygenase.
heart. Hence, it is conceivable that nicotinamide is the primary source of cardiac NAD+ under physiological con-ditions, especially because nicotinamide is the most abun-dant NAD+ precursor in the circulation (2000 nmol/L of nicotinamide versus 7 nmol/L of NR).21,22 In support of this notion, acute NR administration fails to increase cardiac NAD+ content in healthy mice, contrasting with its ability to enhance hepatic NAD+ levels.23 By contrast, dilated car-diomyopathy (DCM) is associated with reduced NAMPT expression and NMRK upregulation, and thus, NR effec-tively replenishes cardiac NAD+.16,24,25
NAD+ Transport and Delivery
NAD+ cannot cross the plasma membrane by passive dif-fusion because of its hydrophilicity, positive charge, and molecular size. Therefore, mammalian cells import NAD+ precursors for intracellular NAD+ synthesis.26 Among these, nicotinamide and NA are the smallest and most membrane-permeant molecules.26–28 NR is transported into cells through the equilibrative nucleoside trans-porter family members.29 As for NMN, initial evidence suggested that it must be dephosphorylated to NR by NT5E (ecto-5’-nucleotidase, best known as CD73) be-fore entering the cell. In fact, deletion of NMRKs, which are required for NR conversion to NAD+, limits the abil-ity of NMN to elevate intracellular NAD+ levels.21,30 More recently, however, the cation/chloride cotransporter SLC12A8 has been recognized as a specific (intesti-nal) NMN transporter.31 Hence, future in vivo studies are required to further clarify the transport mechanisms of NAD+ and its precursors in the card iovascular system, including the recently discovered mammalian mitochon-drial NAD+ transporter, SLC25A51/MCART1.32–34
Another area of intense investigation concerns the biochemical conversions that NAD+ precursors undergo in vivo before they reach the heart or other peripheral tissues. Recent reports suggest that orally administered NAD+ precursors are subjected to extensive first-pass metabolism in the gastrointestinal tract, liver, and later on in the circulation.22,35 Stable isotope tracing revealed that both nicotinamide and NR are converted by the gut microbiota into NA, which is subsequently incorporated into the intestinal and hepatic NAD+ pools through the deamidated pathway.35 In fact, detectable NAD+ metab-olites in the circulation appeared to be generated from both the deamidated and amidated pathways, indicating that oral nicotinamide and NR supplementation stimulates both pathways in vivo. Another mouse study reported that orally delivered NR and NMN are almost entirely con-verted to nicotinamide before reaching the circulation.22 Other studies also reported that intraperitoneal injection of NR and NMN is followed by a surge in the circulating levels of nicotinamide.21,23,36 Even intravenous administra-tion of NR and NMN results within minutes in a substan-tial increase in nicotinamide plasma levels, suggesting an instantaneous conversion of NR and NMN into nicotin-amide within the extracellular space.22 In support of this notion, NR is quickly degraded to nicotinamide when added to murine plasma in vitro,21 whereas intravenously injected NMN is barely detectable in the circulation.22 Although these findings indicate that different NAD+ precursors are converted into nicotinamide, available evi-dence argues against the idea that all these molecules exert identical effects. For instance, as opposed to nico-tinamide, NR efficiently restores NAD+ levels and cardiac function in genetic models of DCM.24 On the flip side, NR failed to improve human skeletal muscle mitochondrial defects,37 whereas NA convincingly restored NAD+ levels and improved mitochondrial myopathy in patients.38
Taken together, the metabolism of NAD+ precursors appears to be much more complex than initially antici-pated, especially in extrahepatic tissues. Hence, system-atic studies should perform head-to-head comparisons of different NAD+ precursors with respect to their galenic and disease-specific properties.
NAD+ Consumption
Steady-state levels of NAD+ are determined not only by its biosynthesis but also by the rate of its utilization. Three classes of enzymes, SIRTs (the sirtuin family of deacetylases), PARPs (poly(ADP-ribose) polymerases), and cADPR (cyclic ADP-ribose) synthases, are known to consume NAD+, resulting in its net catabolism to nicotin-amide (Figure 3).
Mammalian SIRTs are composed of a family of 7 mem-bers (SIRT1–7), which are operative in different cellular compartments, including the nucleus (SIRT1, SIRT6, and SIRT7), cytoplasm (SIRT2), and mitochondria (SIRT3, SIRT4, and SIRT5). Sirtuins are energy sensors that use NAD+ as a cosubstrate to regulate cellular metabolism.39 However, the Michaelis constant values of different sirtu-ins vary significantly,2 indicating that NAD+ concentration does not equally affect the activity of different sirtuins. Indeed, SIRT2, SIRT4, SIRT5, and SIRT6 operate at sub-physiological Michaelis constant values, and thus, their activity is not rate-limited by NAD+, whereas SIRT1 and SIRT3 are highly dependent on NAD+ bioavailability.2 In fact, available evidence indicates that NAD+-replenishing interventions mediate many of their effects in the car-diovascular system through an increase in the activity of SIRT1 and SIRT3.40
At variance with sirtuins, all PARPs are active at rather low levels of NAD+; however, under conditions of increased DNA damage, PARPs may consume sig-nificant amounts of NAD+, which then become rate-lim-iting.22,41 The PARP family is composed of 16 enzymes in mice and 17 in humans, among which PARP1 and PARP2 are considered key DNA damage responders, and thus are required for DNA repair and stability. In con-trast, little is known about the function of other PARP family members, as well as their contribution to global or compartment-specific NAD+ consumption.
Another class of NAD+-consuming enzymes is con-stituted by the cyclic ADP-ribose synthases, including CD38 and its homolog BST1 (bone marrow stromal cell antigen 1, best known as CD157). These ectoenzymes are mainly expressed by immune cells and apparently consume significant amounts of NAD+ to the extent that they are also referred to as NADases.42 Although under normal conditions, high expression of CD38 is only evident in tissues with abundant immune cell populations (Figure 1B), aging and pathological condi-tions associated with elevated immune cell infiltration cause higher expression of CD38 in various tissues.43,44 Notably, cADPR—the product of NAD+ consumption by CD38—is known to regulate calcium homeostasis, and thus might affect cardiomyocyte excitation-contraction coupling.45
Taken together, different enzymes consume NAD+ at different rates and in a cell type- and context-dependent fashion. Although most of the NAD+-using enzymes are expressed in the heart (Figure 1B), their relative contri-bution to net cardiac NAD+ catabolism remains to be elucidated. To this end, for a comprehensive overview of the (patho-)physiological cardiovascular role of these enzymes, we refer the readers to excellent reviews focus-ing on sirtuins,14,40,46,47 PARPs,48,49 or CD38.50
NAD+ AND CARDIOVASCULAR RISK FACTORS
Epidemiological as well as preclinical studies suggest that old age and obesity are among the most important factors that erode health at all levels, including in the car-diovascular system.51,52
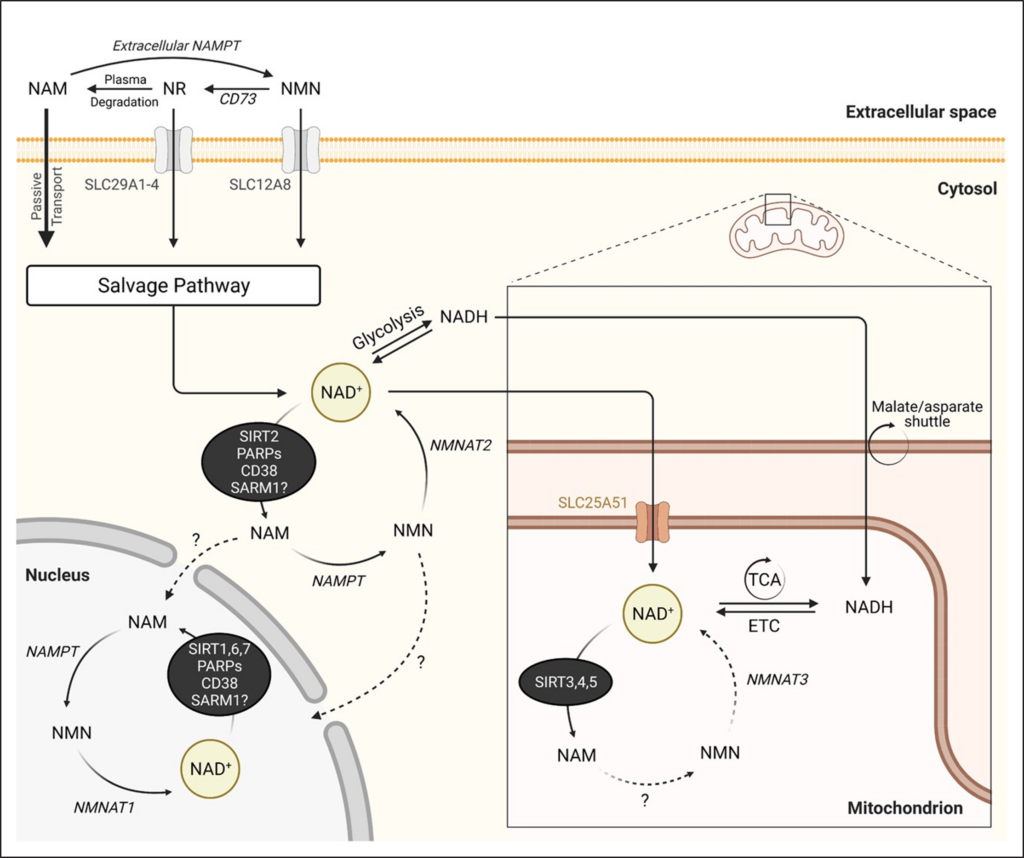
Figure 3. NAD+ catabolic pathways. After their uptake, NAD+ precursors are converted to NAD+ in different subcellular compartments including the mitochondria, cytosol, and nucleus. Accordingly, generated NAD+ controls cellular redox reactions, but also NAD+-dependent enzymes. The latter are responsible for the net catabolism of NAD+ and are composed of 3 classes: SIRTs (the sirtuin family of deacetylases), PARPs (poly(ADP-ribose) polymerases), and the cADPRs (cyclic ADP-ribose) synthases. In addition, SARM1 (sterile α and Toll/interleukin-1 receptor motif-containing 1) has an intrinsic NADase activity, thereby cleaving NAD+ and producing NAM as an end product. ETC indicates electron transport chain; NAD+, nicotinamide adenine dinucleotide, oxidized form; NAM, nicotinamide; NAMPT, nicotinamide phosphoribosyltransferase; NMN, nicotinamide mononucleotide; NMNAT, nicotinamide mononucleotide adenylytransferase; NR, nicotinamide riboside; and TCA, tricarboxylic acid cycle. This figure was created with BioRender.com.
NAD+ Dysregulation in Aging
Intracellular NAD+ concentrations decline with age in var-ious tissues and species, including in humans.10,42–44,53–56 Specifically in the heart, the decline in NAD+ content varies significantly between species and studies, report-ing a 0% to 65% reduction in 2-year-old rodents.57–59 Steady-state NAD+ concentration might decline because of a progressive decay in NAD+ biosynthesis, increased activity of NAD+ degradation enzymes, or a combina-tion of both. On NAD+ biosynthesis, downregulation of NAMPT has been implicated in the age-related decline of NAD+ concentration. However, although this has been documented for multiple tissues/organs,60 it is still un-known whether this also occurs in the heart. Alternatively, the age-related decline in circulating levels of extracel-lular NAMPT that was documented in mice and humans might indirectly affect systemic NAD+ levels.56
A growing body of evidence implicates CD38 as a major culprit in age-related NAD+ decline in mam-mals.42–44 Thus, CD38-deficient aged mice exhibit increased NAD+ content in various tissues.42 Similarly, a specific CD38 inhibitor reverses age-related NAD+ deg-radation and improves several aspects of health, includ-ing cardiac function in aged mice.57 It is interesting that inhibiting CD38 was found to increase NAD+ through an NMN-dependent mechanism, suggesting that in addition to NAD+, NMN is an alternative substrate for CD38.43 Indeed, because of its unique cellular localization with the catalytic site toward the extracellular space, CD38 exhibits an ectoenzymatic activity that degrades circulat-ing NMN in vivo.42 As such, coadministration of NAD+ precursors and CD38 antagonists might be more effi-cacious than CD38 inhibition alone for delaying cardiac aging.57 Because CD38 is predominantly expressed in immune cells,61 it might contribute to a variable extent to the decline of NAD+ content, depending on the abundancy of tissue-resident immune cells.57 Indeed, the progres-sive accumulation of senescent cells that secrete proin-flammatory cytokines has been shown to elevate CD38 tissue levels and hence to promote the age-associated diminution of NAD+ and NMN.43,44 Because murine and human cardiomyocytes are subjected to senescence,62 it is plausible that CD38 might be also involved in the age-related NAD+ decline in the heart, despite the seem-ingly negligible transcriptional expression of CD38 under baseline/healthy conditions (Figure 1B).
PARPs reportedly consume NAD+ to repair age-related DNA damage in aging tissues.63 By contrast, PARP inhi-bition replenishes NAD+ in aged organisms and prevents premature aging caused by deficient DNA repair in sev-eral mouse models.41,64,65 In the myocardium, maintain-ing DNA stability is crucial for normal cellular functions, especially in the postmitotic cardiomyocytes. Given their limited regenerative capacity,66 these long-lived cells are exposed to accumulating metabolic and oxidative dam-age throughout their lifetimes, which ultimately causes DNA damage and PARP activation, thereby reduc-ing NAD+ concentration in the aging heart.58 Activated PARPs might consume significant amounts of NAD+ to fuel the DNA surveillance and repair machinery. However, in the case of genotoxic stress and metabolic collapse caused by excessive DNA damage and NAD+ depletion, respectively, excessive PARP activation might ignite cell death pathways, as reported for a mouse model of heart failure induced by pressure overload.67
Exogenous NAD+ supplementation or overexpression of either NAMPT or NMN adenylyltransferase restores cellular NAD+ levels and prevents cardiac myocyte death in vitro,67 suggesting that increased NAD+ bioavailability or synthesis can counterbalance increased NAD+ con-sumption. In support of this notion, late-in-life dietary intake of nicotinamide delays the hallmarks of cardiac aging in C57BL/6 mice, including reduced cardiac hypertrophy and diastolic dysfunction.15 Along similar lines, oral NMN supplementation to aged mice elicits geroprotective effects on the vasculature by improv-ing aortic stiffness in association with increased arterial SIRT1 activation and reduced vascular oxidative stress.68 Another study reported improved cerebromicrovascular circulation and neurovascular coupling in NMN-treated aged mice.69 Several extracardiac metabolic benefits were also reported for aged mice treated with NMN, nic-otinamide, or NR.9,60,70 Intriguingly, the benefits of NAD+ precursors on mammalian healthspan do not necessar-ily correlate with parallel gains in lifespan.9,71 However, a report suggested that late-in-life NR administration mod-estly extends longevity.10
Taken together, a large body of evidence supports that NAD+-regenerative strategies have a large antiag-ing potential that extends to the cardiovascular system. However, supraphysiological levels of cardiac NAD+ are not necessarily advantageous, because embryonic over-expression of NAMPT reportedly leads to cardiac hyper-trophy in young (6-month-old) mice.72 NAD+ precursors are likely to be beneficial because they avoid the age-related NAD+ overconsumption caused by inflamma-tion (CD38) and DNA damage (PARP1). This assertion has been recently substantiated in an elegant study by McReynolds and colleagues.59 The authors performed NAD+ flux measurements in 25-month-old mice, reveal-ing that a modest and tissue-specific decline in NAD+ is explained by increased NAD+ degradation rather than impaired NAD+ production.59
NAD+ Dysregulation in Obesity
Similar to other cell types, the energy state of cardio-myocytes is reflected by the cellular NAD+/NADH ratio, and the failure to maintain the NAD+ pool is sufficient to cause metabolic imbalance, energy deficit, and func-tional decompensation.73 In this context, it is important to note that the metabolic changes that occur in car-diac NAD+ metabolism depend not only on the severity of the cardiomyopathy but also on the comorbidities, in particular obesity, diabetes, and hypertension.73 A plau-sible explanation for how obesity might induce NAD+ decline resides in the associated subclinical inflamma-tory state.74 Such a sterile (systemic) inflammation might downregulate NAMPT expression, thereby reducing the NAD+ salvage pathway activity in multiple tissues and organs,60 and possibly also the heart. In support of this hypothesis, mice with reduced NAD+ content in their fat depots because of adipocyte-specific NAMPT dele-tion display local (adipose tissue) inflammation but also a severe multiorgan insulin resistance with a 50% re-duction of insulin-induced glucose uptake in the heart, which can be rescued with NMN.75 Consistently, intra-peritoneal injection of NMN stimulates NAD+ biosyn-thesis that can reinstate blood glucose control in obese wild-type mice,60 and in mice with systemic Nampt hap-lodeficiency.76 Other studies using alternative NAD+ precursors, including nicotinamide and NR, have sub-stantiated such metabolic benefits.9,77–79 Intriguingly, nicotinamide-mediated metabolic benefits associate with increased, not decreased, SIRT1 expression.9,79 This observation has been reproduced in the heart,15 and is likely mediated by methyl-nicotinamide–induced inhibition of SIRT1 proteolysis.80
Besides stimulating NAD+ biosynthesis, targeting NAD+ degradation pathways may improve several physi-ological and metabolic aspects of diet-induced obesity. For example, mice with Parp1 or Cd38 deficiency, and mice treated with PARP or CD38 inhibitors, exhibit improved glucose and lipid homeostasis,42,57,64,81,82 as well as exercise capacity.83 Increased NAD+ content in response to these interventions correlates with an increased deacylase activity of SIRT1 and SIRT3 that results in enhanced mitochondrial biogenesis, oxidative phosphorylation, and energy expenditure.42,64 Moreover, the elevation of intracellular NAD+ levels occurring after CD38 deletion protects the mouse heart from high-fat diet (HFD)–induced oxidative stress via activating the SIRT3/FOXO3-mediated antioxidative stress pathway.84
NAD+ Dysregulation in Hypertension
Considering that hypertension is intimately linked to ag-ing and obesity, which both are associated with NAD+ deficiency, NAD+ metabolism has emerged as a poten-tial therapeutic target for hypertension. Indeed, NAMPT expression was found to be downregulated in clinical and experimental hypertension.85 In contrast, systemic NAMPT overexpression protected mice from angio-tensin II–induced hypertension.85 Along the same lines, increased NAD+ biosynthesis upon nicotinamide supple-mentation lowers systolic blood pressure in N-nitro-L-ar-ginine methyl ester–treated mice, eNOS–/– mice, and Dahl salt-sensitive rats.15,86 Although the precise mechanisms underlying such antihypertensive nicotinamide effects are elusive, reduced inflammation has been suggested to be involved.86 Taking into account that nicotinamide is gener-ally regarded safe in humans, it merits further evaluation as an adjuvant therapy of hypertension. Indeed, a recent pilot study showed that supplementation of the alternative NAD+ precursor NR for 6 weeks causes a mild reduction in blood pressure and aortic stiffness in healthy middle-aged and older adults.87 Future trials must examine the effects of NAD+ on patients with hypertension in whom the reduction of blood pressure might be more important.