Graphical abstract
Introduction
All life forms have evolved to cope with varying environments. In humans, malnutrition, infection, or tumorigenesis can perturb access to nutrients, making metabolic flexibility crucial to ensure viability.1 Amino acids are some of the nutrients that show the highest variation in our diet and across tissues.2 Among them, glutamine is the most abundant amino acid in plasma and one of the most highly utilized by cells in culture.3,4 Besides its role as an essential constituent of proteins, glutamine acts as a major contributor to the tricarboxylic acid (TCA) cycle, participating in ATP generation and amino acid synthesis, and to nitrogen metabolism, serving as an amine donor in multiple metabolic reactions, including nucleotide metabolism. The relative contribution of glutamine to the carbon and nitrogen cellular pools, and how they are shaped by local nutrient availability, remains incompletely understood.
While glutamine is classically viewed as a non-essential amino acid, it has long been known that mammalian cells can become dependent on its extracellular availability.4 This phenomenon, referred to as “glutamine addiction,” has been observed both in cell culture and in vivo, and it is thought to result from high proliferation rates exceeding the capacity for intracellular biosynthesis.5 However, it remains unclear whether glutamine addiction results from a scarcity of carbon or nitrogen. Glutamine metabolism plays a crucial role in cancer proliferation, and glutamine addiction is a hallmark of certain cancers,6,7 together with the expression of the oncogene MYC, which encodes the transcription factor c-MYC and induces the upregulation of several transporters and glutamine-related enzymes, including glutaminase.8,9,10,11 Notably, in vivo studies showed remarkable differences in glutamine consumption and abundance across tumor types,12,13,14 increasing the complexity of glutamine addiction in cancer. Understanding which molecular mechanisms and nutrients modulate survival in a glutamine-limited environment could guide novel approaches to limit cancer proliferation.
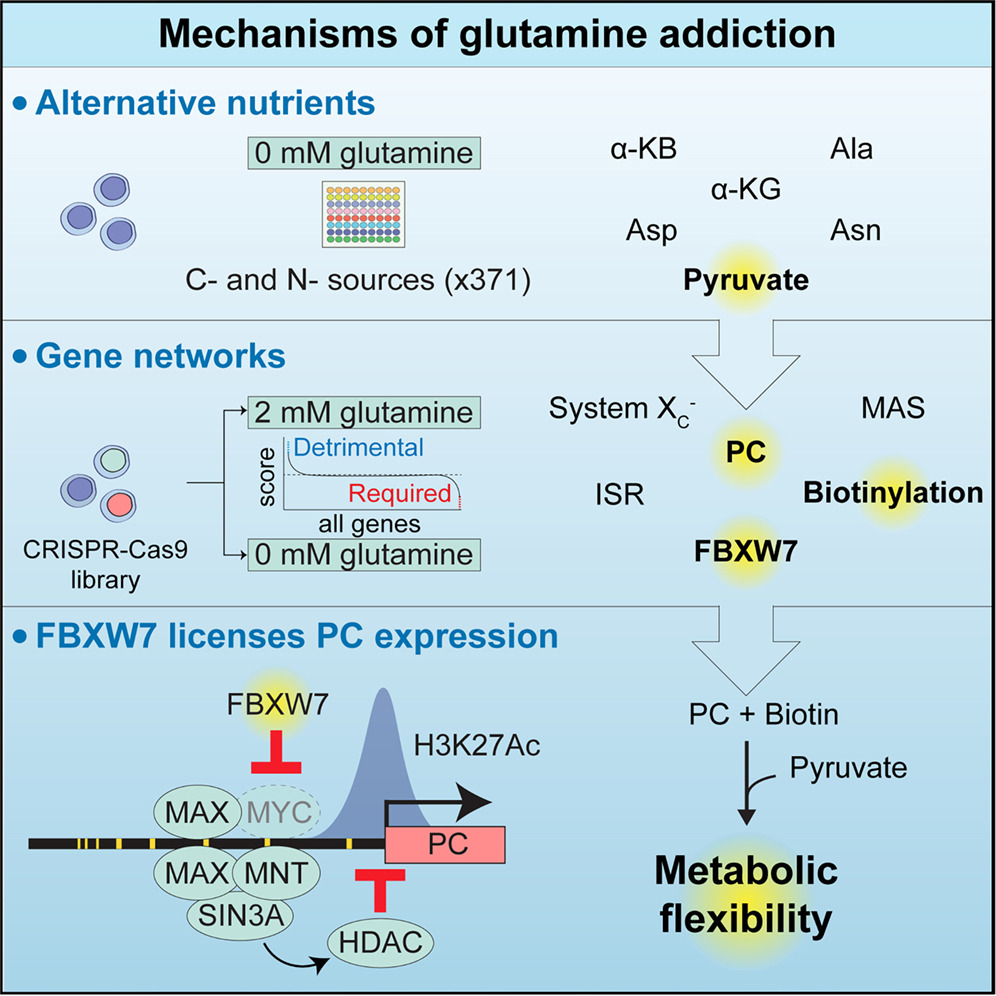
Earlier studies have highlighted metabolic pathways sustaining the proliferation of glutamine-deprived cells, including pyruvate carboxylation, the malate-aspartate shuttle (MAS), and asparagine synthesis,15,16,17 as well as others that become detrimental upon glutamine restriction, such as proline synthesis.18 Despite this important work, we still lack a global, systematic view of the nutrients and pathways involved in cell survival when glutamine is scarce. Here, we combine metabolic tracing with large-scale nutrient and genome-wide genetic screening to provide a unified model of the metabolites and molecular pathways at play in glutamine addiction. We report that biotin licenses proliferation of glutamine-deprived cells by promoting the activity of pyruvate carboxylase (PC), and we further identify an epigenetic mechanism whereby the tumor suppressor FBXW7 regulates glutamine addiction by promoting expression of PC. We show that FBXW7 acts upstream of the MYC extended network, a cluster of transcriptional modulators including repressor proteins that mediate histone deacetylation. We report that FBXW7 depletion leads to c-MYC accumulation, recruitment of MAX, MNT, and SIN3A to the PC promoter, and reduced H3K27 acetylation. Thus, FBXW7 inhibits transcriptional repressor recruitment, licensing PC expression, pyruvate anaplerosis, and proliferation in glutamine-limiting conditions.
Results
Systematic investigation of glutamine addiction
To identify the molecular mechanisms underlying glutamine addiction, we investigated the metabolic reactions that involve glutamine (ChEBI: 28300) and glutamine-derived glutamate (ChEBI: 14321) in humans. Among the 20,420 reviewed protein-coding genes from the UniProt 2025 Reviewed database,19 we found 43 and 127 proteins using glutamine and glutamate, respectively (Figure 1A; Table S1). These proteins mainly encode for metabolic enzymes, transporters, and glutamate receptors, and among them, 28 glutamine-related and 68 glutamate-related proteins are expressed in human K562 myelogenous leukemia cells (transcripts per million, TPM > 1) (Table S1).
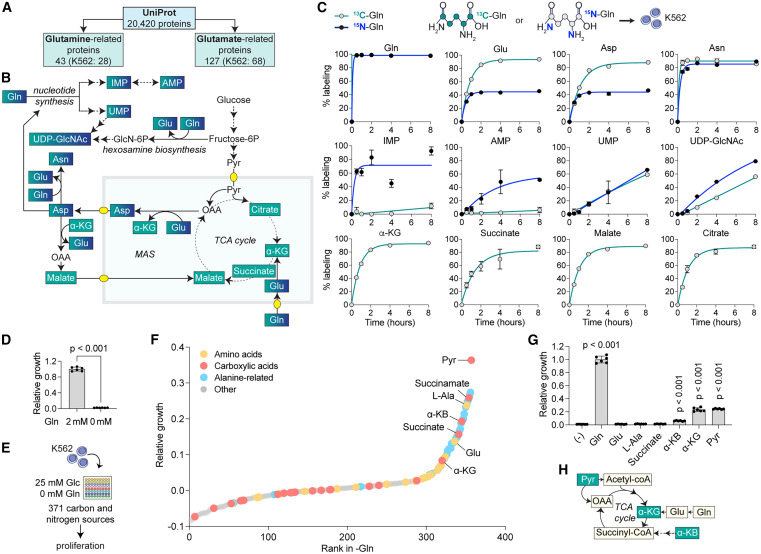
Figure 1 Systematic investigation of glutamine addiction using isotope tracing and nutrient screening
Given the multiple biosynthetic pathways in which glutamine is involved by contributing both carbon and nitrogen groups,20 we performed a time-resolved metabolic tracer analysis in K562 using uniformly labeled 13C-glutamine and 15N-glutamine to measure the participation of glutamine in cellular carbon and nitrogen pools, respectively (Figures 1B, 1C, S1A, and S1B; Table S2). We found that glutamine-derived carbon participates in most of the carbon pools of amino acids, such as glutamate, aspartate, and asparagine, and in glutamate-derived TCA cycle intermediates, including α-ketoglutarate (α-KG), succinate, malate, and citrate, reaching >85% labeling after 8 h. Glutamine-derived carbon was also incorporated at a similar, yet slower rate in pyrimidines (UMP) and pyrimidine-derived metabolites in the hexosamine biosynthetic pathway (UDP-GlcNAc) but not in purines (IMP and AMP), which mostly contain glucose-derived carbons. By contrast, glutamine-derived nitrogen showed a more heterogeneous pattern, and while purines and pyrimidines reached >60% labeling after 8 h, amino acids such as glutamate and aspartate only reached partial 15N-labeling (<50%), indicating significant participation of additional nitrogen sources beyond glutamine. An exception was asparagine, which contains an extra glutamine-derived nitrogen and showed >85% 15N-labeling within 2 h. By contrast, we observed no 13C labeling in pyruvate or acetyl-CoA, suggesting no malic enzyme activity in K562, in contrast to other cells21,22 (Figure S1B).
We next explored whether glutamine addiction could be rescued by the addition of carbon or nitrogen sources. We first tested the ability of K562 to grow in the absence of glutamine and found that it led to a rapid loss of proliferation and viability, indicating strong glutamine addiction (Figures 1D and S1C). We then designed an unbiased nutrient screen comparing the effect of 371 carbon and nitrogen sources, many of which are present in the circulation or in the tumor microenvironment,12,23 on restoring proliferation in glutamine-limited conditions (Figure 1E). We identified a wide range of carbon sources, including carboxylic acids such as pyruvate, α-KG, and α-ketobutyrate (α-KB), some of which had previously been observed in the context of glutamine deprivation,24 glutaminase inhibition,15 and mitochondrial dysfunction25,26 (Figure 1F; Table S3). We also identified metabolites that could act as both carbon and nitrogen donors, including alanine and alanine-containing dipeptides, which can be deaminated to pyruvate by alanine aminotransferase.27 In general, we observed that proliferation increased the most upon addition of carboxylic acids, compared with nitrogen donors (Figures 1G and 1H), consistent with the more limited glutamine contribution to the nitrogen pool in our tracer analysis. Together, our results in K562 indicate that glutamine addiction results mainly from the need for carbon rather than nitrogen.
Anaplerosis, rather than redox imbalance, is the cause of glutamine addiction
To better characterize the metabolome of cells cultured in the presence or absence of glutamine, we performed steady-state metabolomics in glutamine-deprived cells. We found decreased abundance of all TCA cycle intermediates, as well as amino acids such as glutamate, aspartate, and asparagine, which we confirmed using MetaboAnalyst pathway analysis28 (Figure 2A; Table S4). The levels of metabolites related to glycolysis, nucleotide synthesis, the pentose phosphate pathway, and other amino acids showed little to no variation in the absence of glutamine. Based on the key role of carbon supplementation (Figure 1), we investigated whether the addition of the carbon sources identified in our screen could rescue the abundance of metabolites found to be depleted in glutamine-limited conditions. We selected pyruvate as a carboxylic acid that is not directly downstream of glutamine, that does not contain a nitrogen group, and that showed the most beneficial impact on cell proliferation in glutamine-limited conditions in our nutrient screen (Figures 1F–1H). We observed that pyruvate could restore the levels of most metabolites depleted in the absence of glutamine, including TCA cycle intermediates, glutamate, glutamine, aspartate, and asparagine (Figure 2A; Table S4).
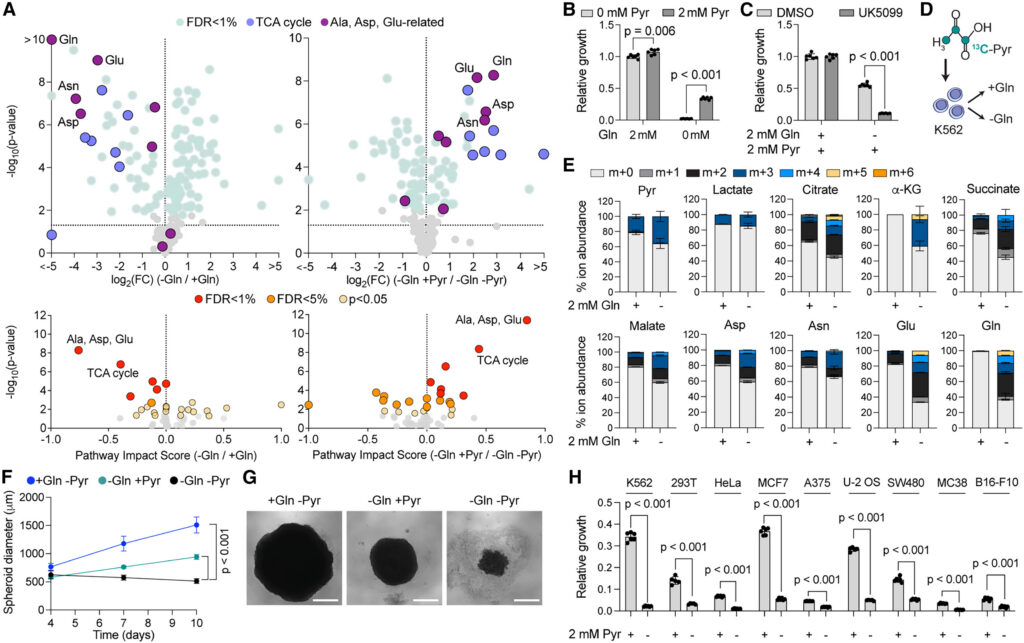
Figure 2 Metabolomics and proliferation analyses of glutamine-deprived cells
These results suggested that pyruvate promotes the proliferation of glutamine-starved cells through anaplerosis, and to exclude a role for exogenous pyruvate in NAD+ regeneration,25,29 we measured the NADH/NAD+ ratio in glutamine-deprived K562 and compared the effect of pyruvate to that of LbNOX, an enzyme prosthetic used as an NADH oxidase.30 We found that glutamine starvation increases the NADH/NAD+ ratio (Figure S2A), in contrast to previous work,31 but as expected from lower TCA cycle activity in glutamine-deprived conditions. We corrected this imbalance by supplementing pyruvate, which performed better than LbNOX in promoting the proliferation of glutamine-starved cells (Figure S2B). Therefore, we conclude that the ability of pyruvate to rescue proliferation under glutamine-deprived conditions is not primarily via restoring NADH/NAD+ balance.
To further validate the anaplerotic route, we supplemented pyruvate to K562 cultured in the absence of glutamine and found that this was sufficient to increase proliferation (Figure 2B). This effect was driven by pyruvate entry into the mitochondria, consistent with the increased abundance of TCA cycle intermediates and confirmed by pharmacological inhibition of the mitochondrial pyruvate carrier (UK5099), which ablated pyruvate-mediated proliferation in glutamine-deprived medium (Figure 2C). We next sought to validate whether pyruvate supports proliferation through anaplerosis and performed a 13C-labeled pyruvate tracer analysis in glutamine-deprived K562 (Figures 2D and 2E; Table S5) and in primary murine CD8+ T cells (Figures S2C and S2D; Table S5). We observed 13C enrichment in most TCA cycle intermediates and TCA-derived amino acids in both glutamine-rich and glutamine-deprived conditions. However, 13C enrichment increased upon glutamine depletion, indicating that cells increase the use of pyruvate for anaplerosis when glutamine is scarce.
To confirm that the role of pyruvate in supporting proliferation of glutamine-starved cells was not cell type-specific, we used HEK239T (293T) spheroids as an alternative 3D model to measure cell proliferation. We found that the addition of pyruvate to the cell culture medium was sufficient to increase proliferation of glutamine-depleted 293T spheroids (Figures 2F and 2G), a result that we further validated across seven additional human and murine cancer cell lines (Figure 2H). Together, our findings confirmed that carbons from pyruvate promote proliferation of glutamine-restricted cells via anaplerosis and that pyruvate-mediated proliferation in glutamine-deprived medium is generalizable across cell lineages, offering a unique opportunity to further study the mechanism of glutamine addiction.
A unified model of glutamine addiction from genome-wide CRISPR-Cas9 screening
We next designed a genome-wide screen aimed at identifying the molecular pathways able to maintain proliferation in glutamine-free, pyruvate-rich conditions. We infected K562 with the Brunello lentiviral genome-wide CRISPR-Cas9 library and transferred cells to a DMEM-based medium containing glucose and pyruvate, either with or without glutamine. After 21 days, we harvested cells and performed next-generation sequencing to determine the abundance of single guide RNAs (sgRNAs) in each condition. We analyzed the data using a Z score-based method26 to calculate the essentiality of each gene, highlighting genes that were required (ΔZ < 0) or detrimental (ΔZ > 0) for the growth of glutamine-starved cells (Figures 3A and S3A; Table S6).
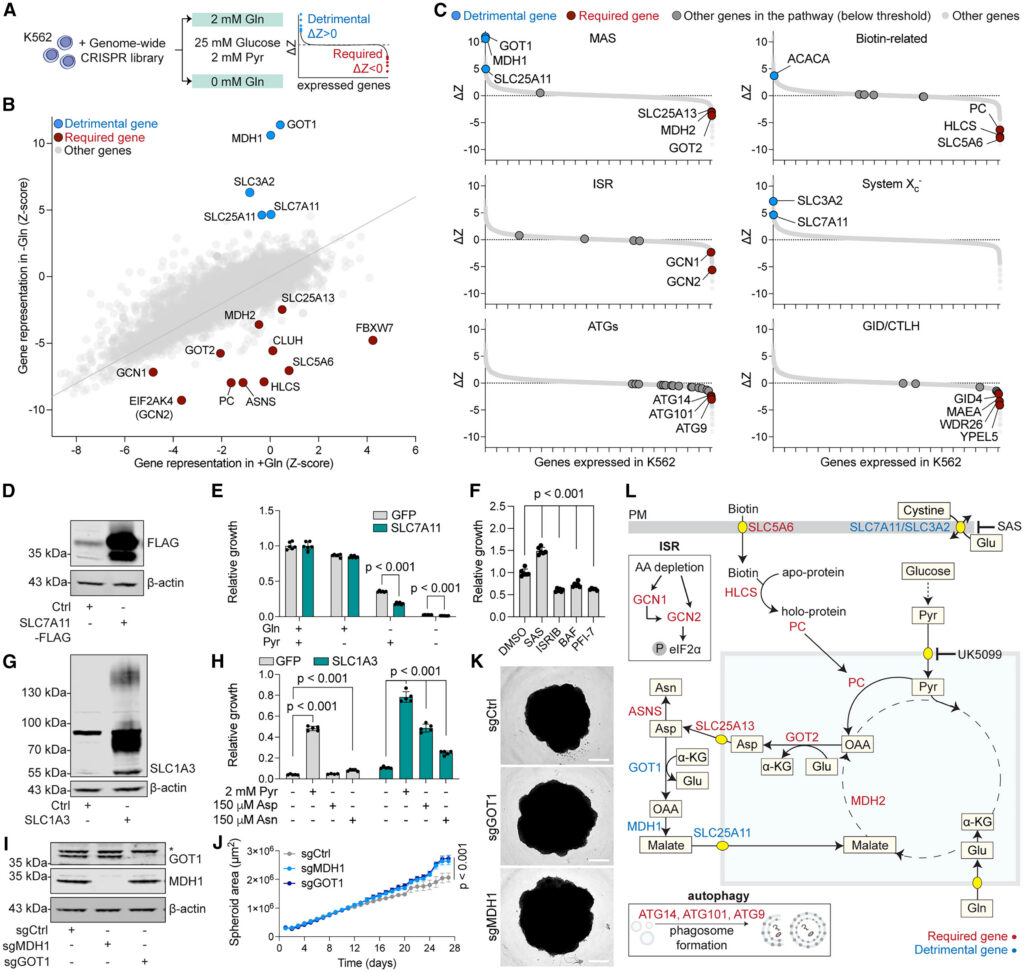
Figure 3 Genome-wide screening provides a unified model of pathways determining glutamine addiction
Validating the robustness of our genome-wide approach, our screen re-identified several factors known to influence the survival of cells in the absence of glutamine, either by promoting or inhibiting proliferation. These include the mitochondrial arm of the MAS24,32 involved in aspartate synthesis (GOT2, MDH2, and SLC25A13); PC, which converts pyruvate to oxaloacetate, thus playing an important role in anaplerosis in cancer cells15,33; the provision of asparagine16,17 via asparagine synthase (ASNS); the amino acid-sensing arm of the integrated stress response (ISR) (GCN1 and GCN2)34; and the plasma membrane cystine-glutamate exchanger system Xc– (SLC3A2 and SLC7A11),35,36 which lies downstream of the ISR37 (Figures 3B and 3C). Our screen also identified individual genes and pathways not previously linked to glutamine addiction, including biotin-related genes (SLC5A6 and HLCS)38,39,40,41; a cytosolic protein involved in the translation of mitochondria-targeted proteins (CLUH)42; autophagy-related genes (ATGs), as well as ubiquitination-related pathways, including the GID/CTLH complex (GID4, MAEA, WDR26, and YPEL5)43,44; and a substrate recognition component of the Skp1-Cullin-F-box (SCF) E3 ligase (FBXW7),45 all of which were required to survive glutamine starvation. Malic enzymes (ME1 and ME2) did not score in our screen, likely because of the exogenous pyruvate available in the medium (Figure S3B).
We validated these hits using genetic and pharmacological tools, first by assessing the contribution of system Xc– and its subunit SLC7A11. Glutamine depletion induced upregulation of ATF4, a known marker of the ISR,34 and of SLC7A11. Notably, we found that pyruvate supplementation partially normalized both ATF4 and SLC7A11 expression in glutamine-deprived cells (Figure S3C). Next, we validated the detrimental role of system Xc– in glutamine-deprived conditions, as highlighted by our screen. We show that overexpression of SLC7A11 decreased growth of glutamine-starved cells (Figures 3D and 3E), while inhibition of system Xc– by sulfasalazine (SAS) promoted proliferation in the same conditions (Figure 3F). These results corroborate previous findings36 and likely hint at a detrimental effect for glutamate export by system Xc– in glutamine-deprived cells.
Next, we used pharmacological inhibition of the ISR (ISRIB), autophagy (bafilomycin [BAF]), and the glucose-induced degradation-deficient (GID), also referred to as the C-terminal to LisH (CTLH) E3 ligase complex (PFI746), and showed that inhibition of these pathways decreased proliferation upon glutamine depletion, despite pyruvate addition, confirming their necessity as highlighted by our screen (Figure 3F). We further corroborated the validity of our model by overexpressing the glutamate-aspartate transporter SLC1A3 and supplementing glutamine-deprived cells with aspartate or asparagine. This approach was previously used to highlight the importance of pyruvate for aspartate synthesis17,29 and for aspartate in promoting proliferation of glutamine-starved cells,24,32 and we confirmed it partially rescued the growth of glutamine-starved K562 (Figures 3G and 3H).
Curiously, the main factors whose depletion promoted the proliferation of cells in glutamine-limited conditions were part of the cytosolic arm of the MAS (GOT1, MDH1, and SLC25A11), which mirrors the mitochondrial arm (GOT2, MDH2, and SLC25A13) by bringing aspartate-derived carbon back to mitochondria. GOT1 and MDH1 are well-characterized for their role in oxidative stress, affecting the growth of cancers such as pancreatic ductal adenocarcinoma.47,48 While higher expression of these genes correlates with poor prognosis in some cancers,49,50 both our screen and validation in 293T spheroids showed that depletion of GOT1 or MDH1 promoted proliferation in glutamine-limited conditions, possibly by preventing re-entry of carbon intermediates in mitochondria and maintaining aspartate in the cytosol (Figures 3I–3K).
Overall, our genetic and nutrient screens highlight genes and nutrients participating in interconnecting pathways and, together with earlier literature, point to a unified model where glutamine-restricted cells rely on pyruvate import into mitochondria, conversion to oxaloacetate, mitochondrial aspartate synthesis, and its export to the cytosol via the MAS (Figure 3L). Intriguingly, we show that blocking the return of aspartate to mitochondria by inhibiting the cytosolic arm of the MAS is beneficial in the context of glutamine restriction and spheroid growth. This further confirms a strong need for cytosolic aspartate, where it can serve as a substrate for both protein and nucleotide synthesis and for asparagine. In addition, our screen also recovered satellite pathways such as the ISR and glutamate export by the system XC– and importantly highlighted biotin metabolism and FBXW7, not previously linked to glutamine addiction.
Biotin metabolism promotes growth in glutamine-limited conditions
Biotin, also known as vitamin B7 or H, is the substrate for biotinylation, a post-translational modification that occurs in mitochondrial carboxylases (PC, MCCC1/MCCC2, and PCCA/PCCB) and in the cytosolic acetyl-CoA carboxylase (ACACA/ACACB). Our screen identified the biotin plasma membrane transporter (SLC5A6) and the biotin conjugating enzyme (HLCS) as required for cell proliferation in the absence of glutamine (Figure 4A). Notably, while human cells encode four classes of biotin-dependent carboxylases, only PC was essential for the growth of cells in glutamine-deprived conditions (Figures 3B and 3C). To validate these hits, we depleted their expression using CRISPR-Cas9 and observed a strong reduction in the ability of K562 to survive glutamine deprivation (Figure 4B). Similarly, we found that K562 cells and 293T spheroids required both biotin and pyruvate for growth (Figures 4C–4E) and confirmed that the depletion of either did not affect PC stability (Figure 4F), pointing to a specific functional requirement for biotinylation.
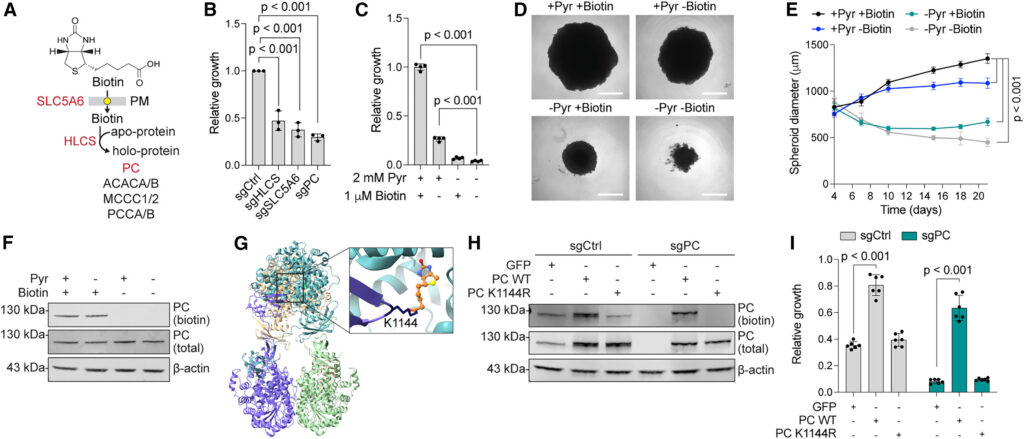
Figure 4 Biotin licenses glutamine-independent growth by promoting PC activity
To test whether PC biotinylation is required for bypassing glutamine addiction, we searched structural databases to identify the biotinylated residue in human PC, which pointed us to lysine 1144 (PDB: 8XL9)51 (Figure 4G). We generated a PC point mutant that cannot be biotinylated (K1144R) and confirmed that wild-type (WT) and K1144R PC were expressed at similar levels and that PC stability was not affected by the lack of biotinylation (Figure 4H). Next, we performed a growth assay to test whether PC biotinylation promoted the proliferation of glutamine-starved cells. While we found that overexpression of WT PC was sufficient to rescue the proliferation of glutamine-starved PC-depleted cells and even promoted the proliferation of PC-competent cells in the same conditions, PC K1144R-expressing cells failed to grow upon glutamine deprivation (Figure 4I). Together, our data indicate that biotinylation of PC is required to support proliferation in glutamine-limited conditions and that biotinylated PC is a limiting factor for the growth of glutamine-addicted cells.
FBXW7 supports PC expression and pyruvate anaplerosis
The gene that scored as the most required factor in the absence of glutamine was the tumor suppressor FBXW7, which, in contrast to most other hits in our genetic screen, is not known to be involved in central carbon metabolism (Figures 3B and S3B). FBXW7 is a well-characterized substrate recognition component for the cytosolic SKP1-CUL1-F-box E3 ubiquitin ligase complex, involved in the degradation of the protein products of oncogenes such as c-MYC, c-JUN, and cyclin E.52,53,54,55
Since a role for FBXW7 in central carbon metabolism was unexpected, we depleted FBXW7 in K562 using CRISPR-Cas9 (Figure 5A) and measured proliferation in glutamine-deprived medium upon supplementation with carbon sources (Figures 5B and 5C). We observed that while FBXW7-depleted cells showed no proliferation defects in a glutamine-rich medium, they grew more slowly in the absence of glutamine, despite the addition of pyruvate. To investigate whether this difference was specifically due to pyruvate metabolism, we supplemented the cell medium with a membrane-permeable α-KG (dimethyl 2-oxoglutarate) analog and found that it was able to promote cell proliferation independently of the expression of FBXW7, pointing to a defect in pyruvate anaplerosis, rather than a global defect in central carbon metabolism. Similarly, FBXW7-depleted 293T spheroids failed to grow in glutamine-free conditions, even after pyruvate supplementation (Figures 5D and 5E). To clarify the fate of pyruvate in FBXW7-depleted cells, we repeated our 13C-pyruvate tracer experiment and focused on malate and aspartate, whose relative fractions of m+3 isotopolog increased in glutamine-limited conditions (Figure 2E). This was indicative of PC-mediated conversion of pyruvate into oxaloacetate, as opposed to m+2 isotopologs, which are generated by pyruvate dehydrogenase (PDH)-mediated conversion of pyruvate into acetyl-CoA. While we confirmed increased enrichment in m+3 isotopologs in the absence of glutamine, we found it to be significantly reduced in FBXW7-depleted cells, irrespective of the presence of glutamine, further suggesting impaired pyruvate anabolism via PC (Figures 5F and 5G; Table S7).
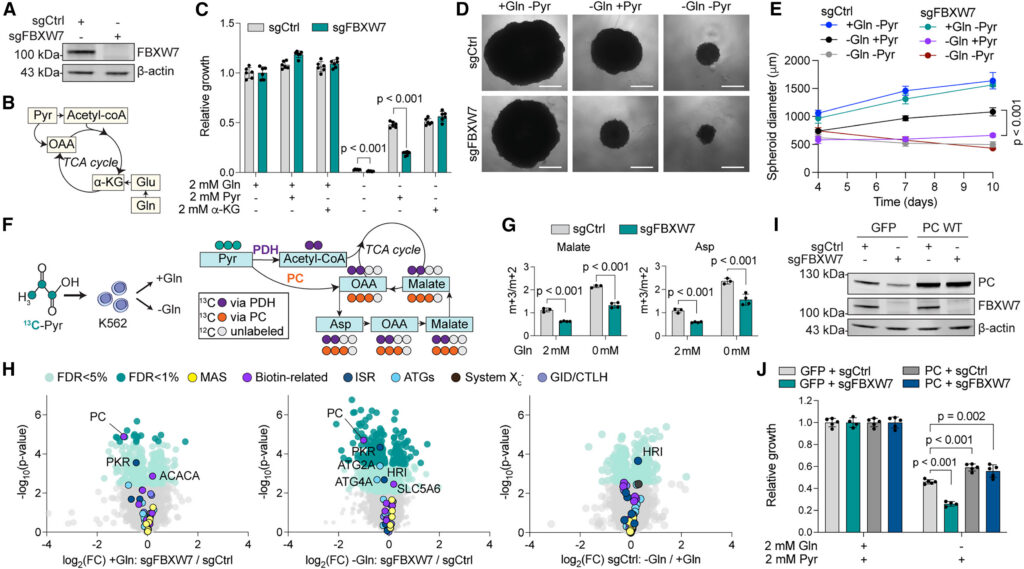
Figure 5 FBXW7 mediates pyruvate anaplerosis by promoting PC expression
As the primary function of FBXW7 is in protein degradation, we performed global cell proteomics to understand how this tumor suppressor and glutamine deprivation affect the proteome. Importantly, we found that PC was among the most significantly decreased proteins in FBXW7-depleted cells, independently of the presence of glutamine, and common to multiple cell lines (Figure 5H; Table S8; Figure S4). Neither FBXW7 nor glutamine depletion affected the protein abundance of any other hits from our screen (Table S8). PC was one of the genes that scored highest among those dictating glutamine addiction (Figures 3B and 3C). Therefore, low PC expression is fully compatible with both the defects in pyruvate metabolism and the impaired growth observed in FBXW7-deficient cells in the absence of glutamine. To test for a causal role, we overexpressed a PC cDNA in FBXW7-depleted cells (Figure 5I). We found that PC overexpression not only conferred a growth advantage to cells during glutamine deprivation, but it also rescued the proliferation of FBXW7-depleted cells to the same extent as in control cells, suggesting an epistatic effect (Figure 5J). Taken together, our results indicate that FBXW7 promotes proliferation in the absence of glutamine by supporting PC expression and subsequently pyruvate anaplerosis.
An FBXW7-MYC-MNT-SIN3A axis regulates PC expression via histone deacetylation
Our observation that PC protein abundance is reduced in the absence of FBXW7 was unexpected given the role of FBXW7 in protein degradation and because these proteins are not localized in the same cellular compartment.56,57,58 To investigate this further, we measured the transcript levels of PC and observed that they were significantly decreased in FBXW7-depleted cells (Figure 6A), mirroring reduced PC protein abundance (Figure 5I). PC mRNA stability was not affected (Figure 6B), suggesting FBXW7 modulates PC expression by regulating factors that affect its transcription.
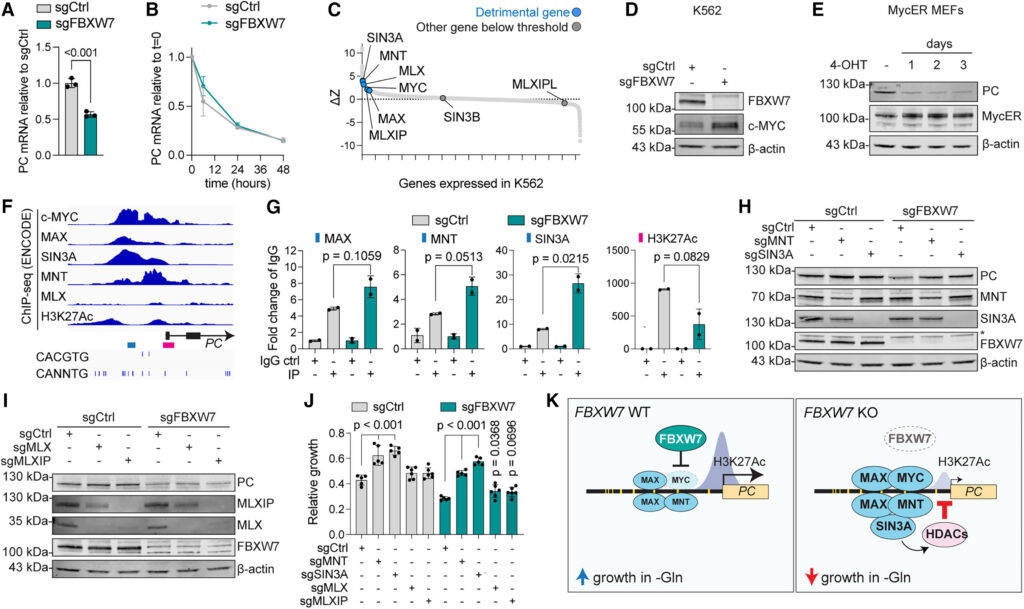
Figure 6 PC expression is repressed by c-MYC and MNT/SIN3A-dependent histone deacetylation
We reasoned that genes involved in PC transcription might also affect proliferation in the absence of glutamine. Thus, we mined our genome-wide CRISPR-Cas9 screen and found a cluster of transcriptional regulators whose depletion was beneficial for growth in the absence of glutamine (Figures 6C and S5A). These included MYC, MAX, MNT, SIN3A, MLX, and MLXIP, known as the MYC extended network.59 These factors bind to both canonical and non-canonical E-boxes to modulate transcription,60 and they can form different heterodimers based on c-MYC accumulation.61 Notably, MNT is known as a c-MYC antagonist62 that competes with c-MYC for MAX binding. MNT recruits the transcriptional repressor SIN3A, which in turn recruits histone deacetylases, thus inhibiting gene expression.63,64
As c-MYC is a target of FBXW7, we first confirmed its accumulation in FBXW7-depleted K562 (Figure 6D). We then focused on the effects of its overexpression, rather than depletion, as MYC is in the top 1% of essential genes in K562.65 To this aim, we employed the MycER mouse embryonic fibroblast line, in which tamoxifen addition initiates c-MYC translocation to the nucleus and DNA binding, a model that has been extensively used to study MYC, including in glutamine addiction.9,18 Importantly, we found that MYC overexpression was sufficient to reduce PC protein abundance (Figure 6E), mirroring the results obtained in FBXW7-depleted K562 and supporting a repressive role for MYC on PC expression.
Next, we focused on the network of transcriptional inhibitors highlighted by our glutamine-sensitized screen. MNT and SIN3A can act as transcriptional repressors by binding genomic DNA and recruiting histone deacetylases (HDACs) to lysine 27 of histone 3 (H3K27).63,64 We mined existing databases to probe for binding of these transcriptional repressors to the PC promoter, as well as for H3K27 acetylation (H3K27Ac), one of the main epigenetic alterations in cancers driven by FBXW7 loss of function.66 We first analyzed chromatin immunoprecipitation (ChIP) and sequencing data from the Encyclopedia of DNA Elements (ENCODE) project67 in K562. We observed a clear signature for recruitment of c-MYC, MAX, MNT, and SIN3A on the PC promoter, as well as H3K27Ac peaks, all in the vicinity of E-boxes (Figure 6F). MLX showed no or little binding, and MLXIP ChIP-seq data were not available for K562. Notably, none of these factors changed in protein abundance following FBXW7 or glutamine depletion (Table S8; Figure S5B). We next performed ChIP in control and FBXW7-deficient cells and measured binding of transcriptional modulators (MAX, MNT, and SIN3A) as well as H3K27Ac at the PC promoter. Importantly, we found that FBXW7 depletion increased binding of MAX, MNT, and SIN3A to the PC promoter and reduced acetylation of the downstream H3K27 site (Figure 6G), suggesting a role for these transcriptional repressors in controlling PC expression.
To test this hypothesis, we used CRISPR-Cas9 to individually deplete MNT, SIN3A, MLX, and MLXIP in both control and FBXW7-deficient cells. Importantly, we observed that depletion of the transcriptional repressors MNT or SIN3A fully rescued both PC expression and proliferation in glutamine-deprived medium in an FBXW7-deficient background, indicating epistasis (Figures 6H and 6J). By contrast, MLX and MLXIP, which showed a less significant score in our screen (Figure 6C, Table S6), did not affect PC protein expression but provided a statistically significant yet minor rescue in glutamine-starved conditions (Figures 6I and 6J). Taken together, our data indicate that upon increased c-MYC expression in FBXW7-depleted cells, a cluster of transcriptional repressors recruits HDACs on the PC promoter, leading to decreased local H3K27 acetylation, lower PC expression, impaired pyruvate anaplerosis, and glutamine dependency (Figure 6K).
Engineered cancer-associated mutations in FBXW7 lead to glutamine addiction
Finally, we aimed to determine the clinical relevance of our model using cancer-associated FBXW7 mutations. To this end, we mined protein structure (PDB: 7D1Y)68 and cancer mutation (Catalogue of Somatic Mutations in Cancer, COSMIC) databases and identified R465, R505, and R479 as the most frequently mutated residues in FBXW7-derived tumors.69 These mutants are predominantly located in the substrate-binding pocket, affecting FBXW7-mediated recognition of target proteins, such as c-MYC (Figure 7A). Notably, the large majority of patient-associated FBXW7 mutations consist of missense substitutions altering the charge of residues in the substrate-binding pocket (R465H, R465C, R505C, and R479Q) (Figures 7B and 7C). To test whether these loss-of-function mutants would mimic the phenotypes we observed in FBXW7-depleted cells, we overexpressed WT and mutant FBXW7 (R465C, R505C, and R479Q) in either an FBXW7-depleted or WT background. Strikingly, overexpression of all FBXW7 mutants led to increased stabilization of c-MYC, reduced expression of PC, and inhibited proliferation in glutamine-deprived medium despite pyruvate supplementation (Figure 7D). These effects were apparent even in WT cells, mirroring the dominant negative effect of FBXW7 mutations in patients69 and linking them to glutamine addiction.
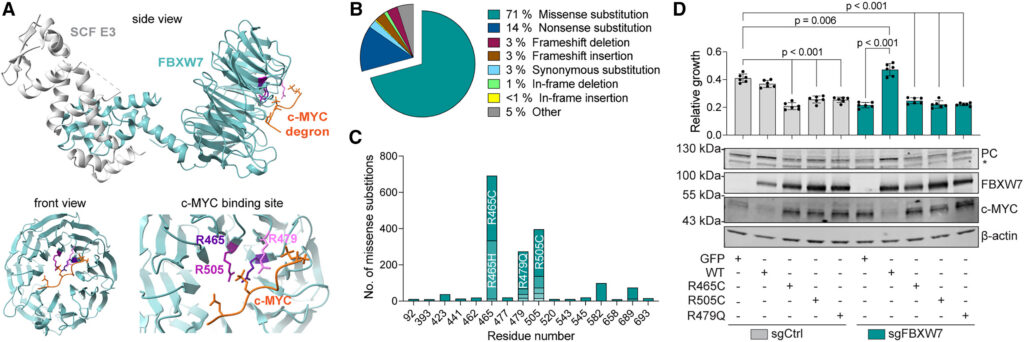
Figure 7 Cancer-associated mutations in FBXW7 lead to reduced PC expression and glutamine addiction
Discussion
Here, we employed parallel nutrient and genetic screening to provide a comprehensive inventory of metabolites and molecular pathways required for proliferation in glutamine-deprived environments. Our systematic approach confirmed the results of previous work that identified metabolites that compensate for glutamine depletion, including carboxylic acids, aspartate, and asparagine.15,16,17,24 In addition, we provide a direct comparison between hundreds of nutrients, covering most of the diversity found in plasma and in tumor interstitial fluid.2,12 Our data highlight carbon-only molecules such as pyruvate and TCA cycle-related metabolites as the main determinants for cell proliferation in the absence of glutamine. While glutamine is both a carbon and a nitrogen source, previous and our findings indicate that its involvement in central carbon metabolism outweighs its role as a nitrogen donor,5 possibly due to other nitrogen-containing molecules, such as amino acids, present in the environment. We confirmed the role of pyruvate in carbon metabolism using a 13C-labeled tracer, showing significant incorporation of pyruvate-derived carbon into TCA cycle intermediates as well as in aspartate and asparagine. In agreement with earlier studies, we showed that aspartate or asparagine supplementation was sufficient to restore proliferation in glutamine-restricted cells.16,17,24
We built on the observation that pyruvate-supplemented cells could survive glutamine starvation to design a genome-wide CRISPR-Cas9 screen to identify genes involved in the proliferation of glutamine-restricted cells upon pyruvate supplementation. Integrating data from our nutrient and genetic screens, 13C-pyruvate tracer, and earlier literature, we propose a unified model in which pyruvate licenses proliferation in glutamine-limiting conditions by serving as an anaplerotic substrate for PC, thus promoting mitochondrial aspartate biogenesis as well as export to the cytosol via the MAS, contributing to asparagine, protein, and nucleotide synthesis. In support of this model, we showed that depleting the mitochondrial arm of the MAS (aspartate export) is detrimental in glutamine-limited conditions, while depleting its cytosolic arm (aspartate import into mitochondria) is beneficial. We also report that in glutamine-deprived cells the amino acid deprivation-sensing arm of the ISR is required for survival, while its downstream effector SLC7A11 and its binding partner SLC3A2 are detrimental. As these proteins form the plasma membrane cystine/glutamate exchanger system Xc–, this could be explained by conservation of intracellular glutamate. Our findings are supported by previous work36 and warrant caution in the use of the system Xc– inhibitors currently being developed for cancer therapy, as they could promote cell survival in a glutamine-limited tumor microenvironment. Importantly, our results also highlight the need for the vitamin biotin and for biotin import and conjugation to proteins, providing an essential cofactor for PC. Together, our findings extend previous work and provide a comprehensive resource identifying both the metabolites and the genes modulating survival of glutamine-starved cells.
Our analysis highlighted the tumor suppressor gene FBXW7 as a central regulator of pyruvate metabolism. FBXW7 is one of the most highly mutated genes in cancers70,71 and degrades the protein product of several proto-oncogenes, including c-MYC, c-JUN, and cyclin E.52,53,54,55 FBXW7 also influences epigenetic remodeling via H3K27 acetylation and tri-methylation and by acting upstream of c-MYC.66 While FBXW7 has been linked to oxidative and liver metabolism,72,73 the mechanism behind these observations remains unclear. We report that FBXW7 mediates anaplerosis by promoting the expression of PC. FBXW7 depletion reduces proliferation in glutamine-deprived environments, independently of pyruvate supplementation. This leads to decreased carbon flux by inhibiting pyruvate utilization in the TCA cycle, limiting aspartate and asparagine synthesis. We highlight PC as the limiting factor for the metabolic flexibility of FBXW7-deficient cells, thus leading to glutamine addiction.
We confirmed c-MYC accumulation upon FBXW7 depletion and report that c-MYC upregulation is sufficient to inhibit PC expression in WT cells. While c-MYC is classically viewed as a transcriptional promoter, its modulation of gene expression is achieved in concert with a network of transcriptional regulators known as the MYC extended network (Figure S5A).59 This includes the c-MYC-binding partner MAX, as well as MNT, MLX, and MLXIP. These proteins form heterodimers that bind E-boxes, either cooperating with or competing against c-MYC-dependent transcription.59 While MLX/MLXIP were suggested to affect c-MYC-dependent metabolic pathways,74 the role of MNT in metabolism remains to be explored. By competing for MAX, MNT acts as a c-MYC antagonist61,62 and inhibits gene expression by recruiting the transcriptional repressor SIN3A and subsequently histone deacetylases.63,64 c-MYC/MAX and MNT/MAX heterodimers can co-exist in cells, but MNT-mediated repression can be dominant over c-MYC transcriptional activation.61,75 We mined the ENCODE database and showed that c-MYC, MAX, MNT, and SIN3A bind the PC promoter upstream of an H3K27 acetylation peak. The PC promoter is characterized by the presence of multiple E-boxes, suggesting complex binding by the MYC extended network. We show that FBXW7 depletion increases recruitment of MAX, MNT, and SIN3A to the PC promoter and decreases H3K27 acetylation. This inhibits PC expression, consistent with a dominant role for MNT/MAX heterodimers in transcriptional repression despite c-MYC accumulation in FBXW7-deficient cells.
c-MYC is a known driver of glutamine addiction that promotes expression of glutaminase and glutamine transporters.8,9,10,11,22 Despite c-MYC accumulation and glutamine dependency of K562, our proteomics analysis of FBXW7-depleted cells did not identify altered expression of these glutamine-related genes, suggesting cell-specific differences. Rather, we observed reduced PC levels across seven FBXW7-depleted human cell lines and showed that PC overexpression restores growth of FBXW7-deficient cells, indicating that PC is both necessary and sufficient to promote proliferation upon glutamine depletion. Our findings provide a better understanding of metabolic pathways regulated by FBXW7 and identify PC as the main downstream effector of FBXW7/c-MYC in glutamine addiction. Interestingly, while some hotspot mutations in FBXW7 share a similar distribution pattern across affected tissues, others appear to be tissue-specific69 and could reflect the ability of FBXW7 mutations to impair substrate binding and inhibit proliferation in glutamine-limited environments. Furthermore, our results suggest that c-MYC upregulation is sufficient to decrease PC expression even in WT cells, highlighting pyruvate carboxylation as a metabolic vulnerability to be targeted for cancer therapy.
An interesting aspect of our findings is the decrease in PC expression, and thus glutamine addiction, in cells overexpressing MYC. We propose two physiological conditions in which this mechanism could be relevant for proliferation. First, MYC coordinates metabolic reprogramming during physiological processes such as pluripotent stem cell differentiation76,77 and hematopoiesis.78 PC repression could limit the role of pyruvate in anaplerosis to promote its use in redox balance via conversion to lactate, thereby enforcing a glycolytic, biomass-producing state that relies on glutamine to replenish the TCA cycle. Secondly, the dependence on glutamine may act as an intrinsic nutrient checkpoint: MYC-driven proliferation proceeds efficiently only when key nutrients, including glutamine, are abundant, providing a built-in safeguard mechanism that would halt uncontrolled cell proliferation in an environment unable to sustain it. Notably, pyruvate import via the mitochondrial pyruvate carrier plays a role in cell differentiation.79,80 We speculate that altering the carbon entry point into the TCA cycle by modulating PC and PDH activity could be a regulatory node in cells with high MYC expression to modulate differentiation and proliferation.
Altogether, our study highlights the power of nutrient-based screening coupled with genetic perturbation strategies to provide a better understanding of the metabolic flexibility of mammalian cells facing nutrient deprivation. We provide a unified model of the molecular mechanisms of glutamine addiction, an important resource for further investigations in the field. Nutrient fluctuations, including glutamine scarcity, can challenge the growth of mammalian cells in different physiological states, such as tissue migration, cell growth, and differentiation. Investigating how cells cope with such heterogeneous conditions will increase our knowledge of the factors supporting metabolic flexibility and could provide applications in pathological settings, deepening our understanding of how cancer initiation and progression can be shaped by glutamine addiction.
Limitations of the study
We systematically analyzed nutrients and molecular pathways modulating the proliferation of glutamine-restricted cells. A common limitation of genetic screens performed in a pooled format is cross-feeding (nutrient transfer between cells via the culture medium) and competition between cells in the population. While we validated many of our hits, future work should include continuing the experimental validation of genes and metabolites highlighted in our screens. Moreover, as we targeted one gene at a time in our assays, it remains possible that compensatory mechanisms and gene redundancy might have masked the involvement of some genes in glutamine-related pathways.
We validated our results in several human and murine cancer cell lines grown in two or three dimensions, as well as in primary cells. In the future, it will be important to further validate our findings in cancer models. We also assigned a role for FBXW7 in PC regulation and pyruvate metabolism: while we focused on the MYC extended network, including MNT and SIN3A, both FBXW7 and c-MYC have a wide range of targets. We do not exclude that additional layers of regulation may exist downstream of FBXW7 and upstream of PC that could contribute to pyruvate anaplerosis and cell proliferation in glutamine-deprived conditions.